



林俊山医生的科普号
- 精选 儿童神经母细胞瘤诊疗专家共识
2017年10月中华小儿外科杂志发表儿童神经母细胞瘤诊疗专家共识神经母细胞瘤(neuroblastoma,NB)起源于肾上腺髓质或椎旁交感神经系统,是儿童期最常见的颅外实体瘤,美国NCI调查结果显示神经母细胞瘤的发病率1975年至2009年为10.54/1 000 000(15岁以下的儿童)。神经母细胞瘤是异质性非常强的肿瘤,一些肿瘤可不经治疗自发消退,但大部分肿瘤发病隐匿,诊断时已出现全身转移并快速进展以致最终致命。国际上经过30余年的多中心协作使得神经母细胞的5年生存率从1974年至1989年的46%上升到1999年至2004年的71%,相对于国际先进水平,我国神经母细胞瘤的诊断、治疗还存在差距,中国抗癌协会小儿肿瘤专业委员会、中华医学会小儿外科专业委员会肿瘤学组为规范我国儿童神经母细胞瘤的诊断和治疗,改善预后,对儿童神经母细胞瘤提出诊疗建议。一、适应证1.未治NB,<18岁;2.治疗前必须明确诊断及分期;3.无严重脏器功能不全。二、治疗前检查1.确诊检查(1)病理检查:肿块切除,切开活检或穿刺活检;(2)骨髓涂片或活检、基于GD2免疫细胞学检测;(3)24 h尿VMA或HVA定量(必要时留置导尿);(4)影像学依据;(5)血NSE。2.分期检查(1)胸部CT增强;(2)腹部及盆腔增强CT,B型超声;(3)眼球B型超声(选择性);(4)ECT全身骨扫描;(5)髂后骨髓涂片+MD检测;(6)MRI;(7)PET-CT(选择性)。3.基因分子检测(1)N—MYC扩增倍数;(2)DNA倍性;(3)1p缺失(选择性);(4)11q缺失(选择性)。4.各脏器功能检查(1)全血象;(2)肝肾功能,电解质;(3)血清LDH;(4)EEG,EKG;(5)流病检测;(6)听力检查。三、基于上述检查可以获得1.明确的组织学或细胞学诊断;2.基于影像学定义的危险因子;3.肿瘤分期及危险度分组。具体诊断标准如下:1.确诊标准(以下两项之一)(1)肿瘤组织光镜下获得肯定的病理学诊断(下列检查可有可无:免疫组织化学染色、电镜检查、血清NSE或尿中儿茶酚胺代谢产物升高);(2)骨髓抽吸涂片和活检发现特征性神经母细胞(小圆细胞,呈巢状或菊花团状排列;抗GD2抗体染色阳性),并且伴有血清NSE或尿中儿茶酚胺代谢产物升高。2.国际神经母细胞瘤病理学分类(INPC)形态学分类(1)神经母细胞瘤(Schwannian间质贫乏):未分化的;弱分化的;分化中的。(2)节细胞神经母细胞瘤,混合型(Schwannian间质丰富)。(3)节细胞神经瘤(Schwannian间质优势):成熟中;成熟型。(4)节细胞神经母细胞瘤,结节型(混合型,Schwannian间质丰富/优势和贫乏)。预后分类(1)预后良好型:<1.5岁,弱分化或分化中的神经母细胞瘤,并且MKI为低度或中度;1.5~5岁,分化中的神经母细胞瘤,并且MKI为低度;节细胞神经母细胞瘤,混合型(Schwannian间质丰富);节细胞神经瘤(Schwannian间质优势)。(2)预后不良型:<1.5岁,未分化的或高度MKI神经母细胞瘤;1.5~5岁,未分化或弱分化神经母细胞瘤,或中度或高度MKI神经母细胞瘤;≥5岁的各种亚型神经母细胞瘤;节细胞神经母细胞瘤,结节型(混合型,Schwannian间质丰富/优势和贫乏)。MKl分为三级:低度(<100/5000);中度(100~200/5000);高度(>200/5000)。3.基于影像学定义的危险因子(IDRFs)(1)单侧病变,延伸到两个间室:颈部一胸腔;胸腔一腹腔;腹腔一盆腔。(2)颈部:肿瘤包绕颈动脉,和/或椎动脉,和/或颈内静脉;肿瘤延伸到颅底;肿瘤压迫气管。(3)颈胸连接处:肿瘤包绕臂丛神经根;肿瘤包绕锁骨下血管,和/或椎动脉,和/或颈动脉;肿瘤压迫气管。(4)胸部:肿瘤包绕胸主动脉和/或主要分支;肿瘤压迫气管和/或主支气管;低位后纵隔肿瘤,侵犯到T9和T12之间肋椎连接处(因为此处易损伤Adamkiewicz动脉)。(5)胸腹连接处:肿瘤包绕主动脉和/或腔静脉。(6)腹部和盆腔:肿瘤侵犯肝门和/或肝十二指肠韧带;肿瘤在肠系膜根部包绕肠系膜上动脉分支;肿瘤包绕腹腔干和/或肠系膜上动脉的起始部;肿瘤侵犯一侧或双侧肾蒂;肿瘤包绕腹主动脉和/或下腔静脉;/10fI瘤包绕髂血管;盆腔肿瘤越过坐骨切迹。(7)椎管内延伸:轴向平面超过1/3的椎管被肿瘤侵入,和/或环脊髓软脑膜间隙消失,和/或脊髓信号异常。(8)临近器官/组织受累:包括心包、横膈、肾脏、肝脏、胰一十二指肠和肠系膜。注:下列情况应当记录,但不作为IDRFs:多发原发灶;胸水,伴有/无恶性细胞;腹水,伴有/无恶性细胞。需要的影像学技术包含:CT和/或MRI;I一123MIBC-;Tc-99m MDP骨扫描。4.INSS分期1:局限性肿瘤,肉眼完全切除,伴有/无镜下残留,同侧与肿瘤非粘连性淋巴结镜下阴性(与原发肿瘤融合粘连并一并切除的淋巴结可以是阳性的)。2A:局限性病变,肉眼不完全切除,同侧与肿瘤非粘连性淋巴结镜下阴性。2B:局限性病变,肉眼完全或不完全切除,同侧与肿瘤非粘连性淋巴结镜下阳性,对侧肿大的淋巴结镜下阴性。3:无法切除的单侧肿瘤越过中线,区域性淋巴结阴性/阳性;单侧肿瘤未超越中线,对侧肿大淋巴结阳性;中线部位肿瘤,通过肿瘤直接侵犯(无法切除)或淋巴结转移方式向两侧延伸。4:任何原发肿瘤伴有远处淋巴结、骨髓、肝、皮肤和/或其他器官(除外4S期)播散。4S:原发肿瘤为局限病变(I、IIA或IIB期),并仅限于皮肤、肝和/或骨髓转移(限于年龄<1岁的婴儿),骨髓微量受累即骨髓穿刺或活检显示神经母细胞占所有有核细胞的比例<10%;如果行MIBG扫描,骨髓必须是阴性的)。注:中线为脊柱,越过中线是指侵犯到或越过脊柱的对侧缘。若存在多发原发病变,按照受累范围最广的病变进行分期。5.危险度分组(COG)低危:①所有1期;②<1岁所有2期;③>1岁MYCN未扩增2期;④>1岁,MYCN虽扩增但INPC为预后良好型2期;⑤MYCN未扩增,INPC为预后良好型且DNA为多倍体4S期。中危:0<1岁,mycn未扩增3期;②>1岁,MYCN未扩增且INPC为预后良好型3期;③<1岁半,MYCN未扩增4期;④MYCN未扩增,DNA为二倍体4S期;⑤MYCN未扩增且INPC为预后良好型4S期。高危:①>1岁,MYCN扩增INPC为预后不良型2期;Q1岁,MY—CN未扩增但INPC为预后不良型3期;④>1岁,MYCN扩增3期;⑤<1岁,mycn扩增4期;⑥>1岁半的所有4期;OMYCN扩增的4S期。儿童神经母细胞瘤的治疗一、治疗计划1.低危:存在影像学定义的危险因子或具有症状(脊髓压迫、肝肿大呼吸窘迫、泌尿及消化道梗阻、严重凝血异常等)的先行化疗治疗。治疗选择:①手术+化疗(化疗至VGPR后4个疗程,一般4~6疗程,总疗程不超过8个疗程):MYCN扩增的Ⅰ,Ⅱ期;大于18个月2B期;INPC为预后不良且DNA为二倍体的2B期;具有临床症状的4s期;②其他情况:手术、术后密切随访(每个月1次)。2.中危:化疗前或化疗中(约4疗程左右)择期手术,术后化疗至VGPR后4个疗程,总疗程不超过8个疗程,必要时行二次手术。维持治疗:13-cis-RA 160 mg/m2,14d/月,共6个月。3.高危:先化疗(约4疗程左右)后择期手术。术后化疗至VGPR后4个疗程,总疗程不超过8个疗程,常规化疗结束后自体干细胞移植和瘤床放疗(推荐行序贯自体干细胞移植,瘤床放疗在两次自体干细胞移植之间进行)。停化疗后13-cis-RA 160mg/m2,14 d/月,共6个月。(若不具备干细胞移植条件可继续进行化疗至12个疗程)二、手术原则1.手术时机如果存在IDRFs中的一项或多项应推迟手术,通过化疗降低手术并发症的危险性后再手术治疗。2.手术范围(1)切检:若初诊患儿无法明确病理诊断,或者穿刺活检获得的组织无法满足基因分子生物学分析,可考虑对原发灶或转移灶进行手术切检。(2)部分切除或完全切除:在保证安全的前提下切除原发灶及区域内转移淋巴结,如果手术带来的并发症不可以接受,则行部分切除,残留部分通过放化疗继续治疗。如果通过化疗使转移灶局限,可行手术切除转移灶,比如肝或肺孤立病灶,颈部转移灶可行广泛淋巴结清扫术。三、放疗适应证(1)所有高危组患儿均需接受原发部位、持续存在的转移灶的放疗。(2)低一中危组出现脊髓压迫症状、呼吸窘迫综合征者化疗反应不够迅速可考虑放疗。(3)中危组病灶进展的。四、疗效评估标准1.完全缓解(complete response,CR):所有原发灶和转移灶消失,儿茶酚胺及代谢产物恢复到正常水平。2.非常好的部分缓解(very good partial response,VGPR):原发灶体积减少90%~99%,所有可测量的转移灶消失,儿茶酚胺及代谢产物恢复到正常,99Tc扫描骨骼病灶可以是阳性(因为骨骼转移灶未愈合),但如果行MIBG检查,所有病灶均阴性。3.部分缓解(partial response,PR):所有原发灶和可测量转移灶体积减少超过50%,骨骼阳性病灶的数目下降超过50%,不超过一处的骨髓阳性部位可以接受。4.混合性反应(mixed response,MR):没有新的病灶,在任何一个或多个可测量的病灶体积下降超过50%,同时存在其他任何一个或多个病灶体积下降小于50%,任何存在的病灶体积增加小于25%。5.无反应(no response,NR):没有新病灶,任何存在的病灶体积下降小于50%或增加小于25%。6.进展性疾病(progressive disease,PD):出现新病灶,已存在可测量的病灶体积增加超过25%,骨髓由阴性转阳性。
林俊山 主任医师 福建医科大学附属第一医院 小儿外科5130人已读 - 案例 腹腔占位
治疗前患者女,6岁,因“反复腹痛伴发热2天余。”于,既往无相似症状。患者夜间以“腹痛”起病,以脐周为甚,呈阵发性闷痛,改变体位无明显缓解,无放射他处,与进食无关,排便后不能缓解。随后就诊于当地医院,期间出现发热,最高达38.4℃,后出现畏冷、寒战,查血象示:WBC19.210^9/L,N%74.3%,CRP:107ng/L,Hb92g/L。查腹部彩超示:腹腔内囊性包块(性质待定),大小约:23.5cm16.5cm0.5cm。腹部CT平扫示“腹腔内不规则类囊样液性密度影”。转诊我院后完善全腹部CT增强提示:腹腔巨大占位性病变(脉管畸形可能,内出血?)。查体:神志淡漠,T:36.2℃,P:121次/分,R:21次/分。HP:100/87mmHg。四肢稍凉,腹肌紧,腹式呼吸运动存在,未见腹壁静脉曲张,未见胃、肠型及异常蠕动波,全腹压痛,可疑反跳痛,以脐周为甚,肝脾未触及,墨菲氏征阴性,麦氏点无压痛,肝区未及叩击痛,双侧肾区未及叩痛,肝浊音界存在,移动性浊音(-),肠鸣音2次/分。入院后予以心电监护、补液扩容、止血、积极术前准备等处理。治疗中术中见:囊肿张力明显,占据了腹腔2/3的大部分。囊肿内含有血性浆液,似乎是多房的。将囊肿小心地抬出腹部,见囊肿的上缘与网膜连续。治疗后治疗后7天术后病理确诊
 林俊山 主任医师 福建医科大学附属第一医院 小儿外科56人已读
林俊山 主任医师 福建医科大学附属第一医院 小儿外科56人已读 - 耳朵前的小洞--耳前瘘管
很多小宝宝一出生,耳朵附近就长了一个小洞 耳前瘘管会遗传吗?耳前瘘管是常染色体显性遗传性疾病,男女都会遗传。国内统计过耳前瘘管发生率高达1.2%,单侧与双侧发病比例为4:1。我们常发现来看病的患者时,其家族里面也会存在耳前有小洞,而且大多数人单侧还是双侧发病与上代也相同,也有少数人会同时伴有腭裂、小耳畸形、副耳等先天性畸形等。 耳前瘘管的分型单纯型耳前瘘管一辈子与主人和睦相处,不会分泌及发炎,因此无需处理。分泌型耳前瘘管常分泌白色乳膏样物,有点臭味,可能伴有瘙痒感,禁止挤压或自行挑破,以防感染。如果影响社交,可考虑手术切除。感染型耳前瘘管经常感染,洞口附近皮肤红肿、疼痛,严重时破溃溢脓。感染型耳前瘘管常在分泌型耳前瘘管合并挤压后产生。耳前瘘管无症状时不需治疗,一旦出现感染则需手术治疗,完全摘除瘘管是防止复发的唯一有效途径。 目前,耳前瘘管的治疗方法1、发生急性感染,或因感染引起皮肤破溃者应先使用抗生素控制炎症反应后再行手术;2、局部有脓肿形成者应先切开引流,待炎症反应控制两周后再行手术;3、一般换药法:局部清创换药至痊愈。耳前瘘管需要注意什么?1、一定不要揉,不能用手常去触碰,避免挤压。2、保持局部清洁。洗脸的时候,记得用清水洗洗耳朵前后,避免感染。3、保持耳部干燥。如果耳朵周围有水,用棉签擦拭就可以,但不能太用力。4、不能用异物戳小洞,发生感染后,不要自行挑破脓肿;5、如果出现痒、痛,要及时看医生。
 邱志欣 医师 福建医科大学附属第一医院 小儿外科160人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科160人已读 - 挺起胸膛--漏斗胸
所谓漏斗胸,就是指患者的前胸壁出现凹陷,呈现出漏斗状的外观,这是一种先天性的渐进式病变,出生时可能已经存在病变,往往随着年纪增长而变得越来越明显。其原因是肋骨生长不协调,过长的肋骨挤压使胸骨向后凹陷,附着于胸骨下端的膈肌中心腱过短,使胸骨和剑突受到向后的牵拉凹陷,宫内受压、呼吸道梗阻、部分膈肌前方肌肉纤维化、胸骨和肋软骨发育障碍、结缔组织异常等。漏斗胸的危害中、重度漏斗胸将导致心脏受压移位,肺也因胸廓畸形而运动受限,影响患者的心肺功能。凹陷的胸壁使胸腔整体容量减小,肺的扩张受到抑制,尤其是吸气时肺扩张受限,阻力增加,易发生上呼吸道感染。其次胸腔减小后,心脏活动受到限制可使其射血量减少,从而运动时供血不足,患儿活动后出现心悸、气促和呼吸困难等症状,甚至发生心力衰竭。心理上患者容易出现自卑、羞耻感、社交障碍、自闭等心理问题,且随年龄增加,胸廓凹陷畸形逐渐明显,影响美观,严重影响身心健康。 漏斗胸三注意注意一漏斗胸应该要注意积极的治疗,避免病情造成影响;注意二漏斗胸要注意饮食规律,要保证一日三餐定时定量,每天应多补充两次点心,临睡前可以加一次牛奶,每天不宜吃得过饱,要注意营养的搭配,保持营养全面均衡;注意三要注意饮食方面,由于漏斗胸人群消化功能不好,因此,饮食调整速度要慢,要合理的选用食物,多吃含有丰富优质蛋白质以及锌铁和维生素的食物,多吃富含钙质的食物以及绿色蔬菜,能增强抵抗能力。手术治疗目前我科采用的Nuss技术治疗法是一种治疗漏斗胸的新型微创技术,Nuss技术因其切口小、创伤小、出血少、并发症少,不仅可以纠正畸形,而且外观好,有效改善心肺功能,能够重塑患者的自信心。这样的手术并非将畸形骨骼折断后进行的塑形,而是在对畸形骨骼整体不做局部损伤的前提下进行的整体塑形。这种方法相当于在体内置放一个“模具”,使骨骼按照“模具”的形状进行塑形,用“模具”把凹陷的胸骨顶起来,顶起来之后再从另外一边腋下出来,出来以后固定在两边的皮下。
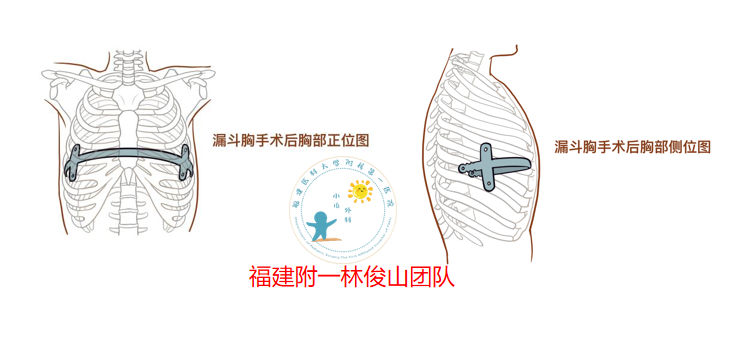 邱志欣 医师 福建医科大学附属第一医院 小儿外科72人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科72人已读 - 这种能迫使男孩子蹲着尿尿的疾病,你知道吗?
今天就来为大家科普一下~这种能迫使男孩子蹲着尿尿的疾病尿道下裂什么是尿道下裂?从小我们就知道男孩和女孩尿尿的方式不一样可有些男孩因为与常人不一样只能蹲着小便……尿道下裂(Hypospadias)是由于前尿道发育不全,胚胎发育过程中尿生殖沟没有自后向前在中线完全闭合,造成尿道口达不到正常位置的阴茎畸形。它属于泌尿生殖系统最常见的先天畸形,在我国,每1000个男孩中约有3人发病。尿道下裂使儿童尿道开口畸形,不能正常排小便,严重影响身心发育。患儿一般伴有阴茎下弯,影响阴茎的正常发育,如不及早治疗,将影响到孩子的心理健康及以后的婚姻、生育。如何判断孩子是否有尿道下裂?一般在出生体检时即可诊断出婴儿尿道开口的位置是否存在异常,家长平时可通过观察孩子的尿道口、阴茎形态以及尿流来判断是否患有尿道下裂。一般情况下,患有尿道下裂的孩子,尿道口不在阴茎顶端,阴茎勃起后向下弯曲,阴茎的包皮分布异常,站立排尿时往往会弄湿裤子。尿道下裂是怎么引起的目前病因尚未明确,已经证实的原因包括:雄激素受体异常、遗传基因突变、内分泌失调、异常细胞间信息传递、表皮生长因子表达降低和环境因素等。近20年,尿道下裂发病率显著上升,可能与环境中广泛存在的雌激素和抗雄激素类物质的污染有关,环境污染物使正常内分泌因素改变而发生畸形。另外,孕期服用避孕药物、低出生体重亦为高风险因素。尿道下裂有轻重程度之分吗?根据尿道外口位置分型,尿道下裂可分为:1.远端型,包括阴茎头、冠状沟、冠状沟下型。2.中间型,即阴茎体型。3.近端型,包括阴茎阴囊型、阴囊型和会阴型。开口位置距离龟头越远,就越严重。尿道下裂治疗方式及预后手术治疗为主要的治疗方法,手术最佳时机一般在3岁左右,手术方式包括阴茎弯曲的矫正及尿道成形。具体手术方式应结合患者年龄、病变类型进行个性化治疗。总体来说尿道下裂合并阴茎下弯手术效果满意,术后孩子能正常站立排尿,外观接近正常,成年后也能进行正常的性生活。
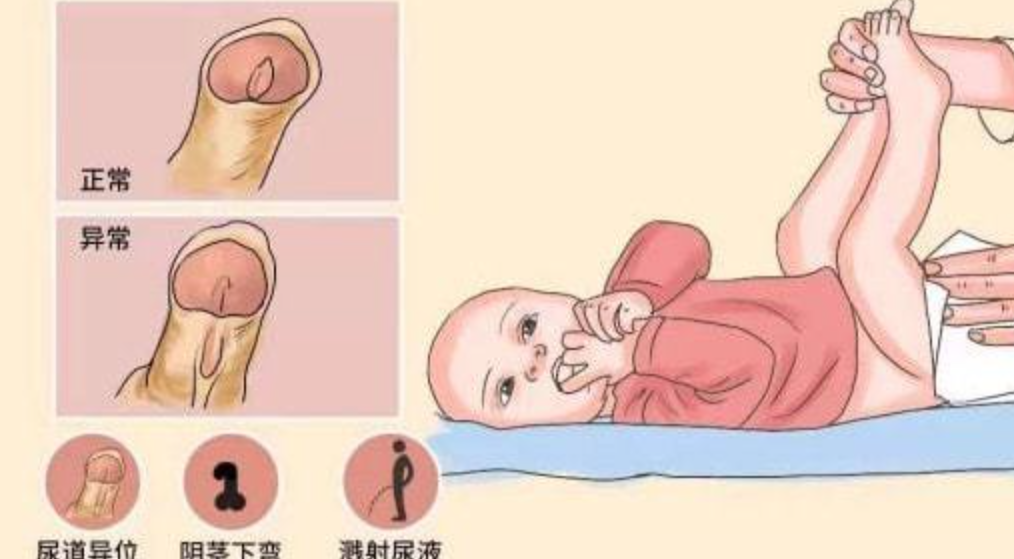 邱志欣 医师 福建医科大学附属第一医院 小儿外科76人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科76人已读 - 惊!刚出生的孩子肚子上居然有个“大包”
大家是不是很好奇,刚出生的宝宝为什么肚子上有个“大包”-脐膨出1什么是“脐膨出”呢?脐膨出又称胚胎性脐带疝,与脐疝不同,它是由于前腹壁的发育缺损,腹腔内脏通过脐部缺损突入脐带基底部,外面覆盖一层薄而透明或半透明的囊膜,没有皮肤附着。2是什么原因造成的呢?胚胎发育异常被认为是本病的主要病因,染色体异常、部分药物的宫内暴露、多胎妊娠及母体肥胖等其他因素也可能导致本病的发生。本病少见,发病率1-2.5/5000个活产婴儿,男:女≈3:2。3怎么去认识这个疾病呢?本病诊断不难,外观即可确诊。主要表现为腹部(脐部)肿物,肿物被半透明或透明膜所覆盖,脐带由其上伸出。有时透过囊膜可见囊内器官:如肠管、肝、脾、肾等。本病伴发其他先天性畸形可能性大,部分伴发畸形需通过检查才能明确,如:先天性心脏病、肠旋转不良、肛门直肠畸形等等,故一旦发现需及时就医。4治疗原则?本病属于危急重疾病,家属需正确处理(无菌塑料膜覆盖囊膜、囊膜保温保湿、患儿保温)后及时送医。医师会根据腹壁缺损大小、是否合并畸形、有无囊膜破裂、有无严重感染等选择最佳治疗方案,可采用手术治疗或非手术治疗。①手术治疗:A:一期修补:腹壁缺损直径<5cmB:二期修补:腹壁缺损直径≥5cm②非手术治疗:结痂剂:适用于不适宜手术者,如:巨大型脐膨出、就诊时间晚、全身情况差、严重感染等预后:存活率为70-95%,大多数死亡与伴发的心肺畸形、染色体异常或严重感染有关。5小结脐膨出不可怕,保护好外露的组织,注意保暖,通畅呼吸,减低腹内压,预防使用抗生素,及时转入有条件的正规医院,为患儿的进一步治疗和最终治愈赢得时间。
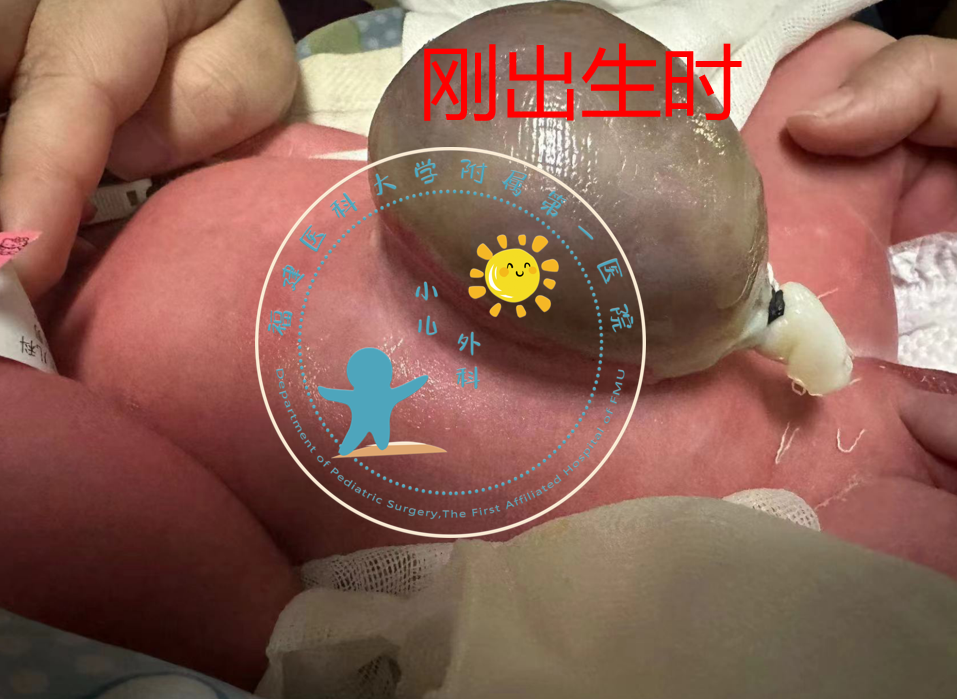 邱志欣 医师 福建医科大学附属第一医院 小儿外科18人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科18人已读 - 图文文章 别让隐睾成隐忧!孩子隐睾怎么治疗?
3岁孩子隐睾,是否要手术?有些家长可能会认为现在孩子还小也不需要这个功能等青春期再说也不迟错!!对于先天性隐睾医生建议出生后6个月后要尽早进行手术且一定要在2周岁前完成3岁小孩发现隐睾若不及时处理,青春期睾丸可能就没有产生精子和产生雄性激素的功能。那么,儿童隐睾具体是怎么回事?怎么发现孩子是不是隐睾?01、儿童隐睾新生儿隐睾发病率为3%-5%,早产儿中发病率约为30%,是正常成熟儿的6倍左右。出生后睾丸仍有自行下降可能,一般在出生后3-6个月。1岁以后睾丸基本不会自行下降。1周岁时发生率为1%。单侧约占75%,双侧约占25%,右侧明显多于左侧。02、儿童隐睾是怎么形成的?孕25-30周时,在睾丸引带的引导下胎儿睾丸下降至腹股沟阴囊,此过程为激素依赖,睾酮被认为是促睾丸下降的动力因素。睾丸下降至阴囊常是先左侧后右侧。如睾丸未降至阴囊底而沿睾丸引带尾端其他分支下降至会阴、耻骨部或股部,则成为异位睾丸。未降入阴囊的睾丸常有不同程度的发育不全,体积明显小于健侧,生殖细胞发育障碍,精原细胞数量减少,位置越高,病损越重;年龄越大,病损越重。单侧隐睾,对侧正常下降也可能有病理性改变(交感性病变)。03、隐睾会有什么危害?睾丸未降及其相关疾病的并发症和后遗症包括腹股沟疝、睾丸扭转、睾丸外伤、生育能力低下和睾丸癌。其中生育能力下降和睾丸癌最为严重。睾丸的生精过程受许多因素的影响,特别是对温度的变化非常敏感。正常情况下,阴囊中的温度低于腹腔温度约1.5℃~2℃,正好适合于精子的产生和雄激素的分泌,如果睾丸留于腹腔中,则精子的产生及雄激素的分泌将受到很大的影响。隐睾的话,睾丸的生精能力很差,精子的成活率也低。所以,即使孩子还没到青春期,这个问题也要及时干预,早发现早处理。04、隐睾的治疗隐睾影响这么大,那要怎么治?关于隐睾的治疗,其目的为保全患者生育能力,避免精神心理不良影响,减少性功能不正常情况,预防并发症的发生。睾丸未降跟激素水平有关,某些医疗中心使用激素治疗诱导睾丸下降,并且可能作为手术治疗的辅助手段继续使用,以期改善生育力。重点来了!我们不建议使用激素治疗因为睾丸下降依靠局部睾酮浓度此浓度远高于全身用药所能达到的浓度手术治疗隐睾是最安全、疗效最佳的治疗方法。一般来说,对于6月龄(矫正胎龄)时睾丸仍未下降至阴囊者,建议尽早手术,最好能在12月龄前手术,建议最晚不超过18月龄。对于回缩睾丸,需每年随访并持续至青春期,直到睾丸不再回缩且停留在阴囊内。从术后的预后来看,经过及时纠正后,单侧隐睾的生育率与正常男孩子基本相同,但双侧隐睾的孩子生育率则显著下降。从长远来看,有过隐睾的孩子,以后发生睾丸生殖细胞肿瘤的风险也将增加5-10倍,尤其是腹腔内隐睾或者双侧隐睾的孩子。而早期给予手术治疗的孩子虽然可在一定程度上降低睾丸癌变的概率,但是发生癌变的概率仍是比正常人要高,因此,隐睾患儿在青春期以后仍需定期复查体检
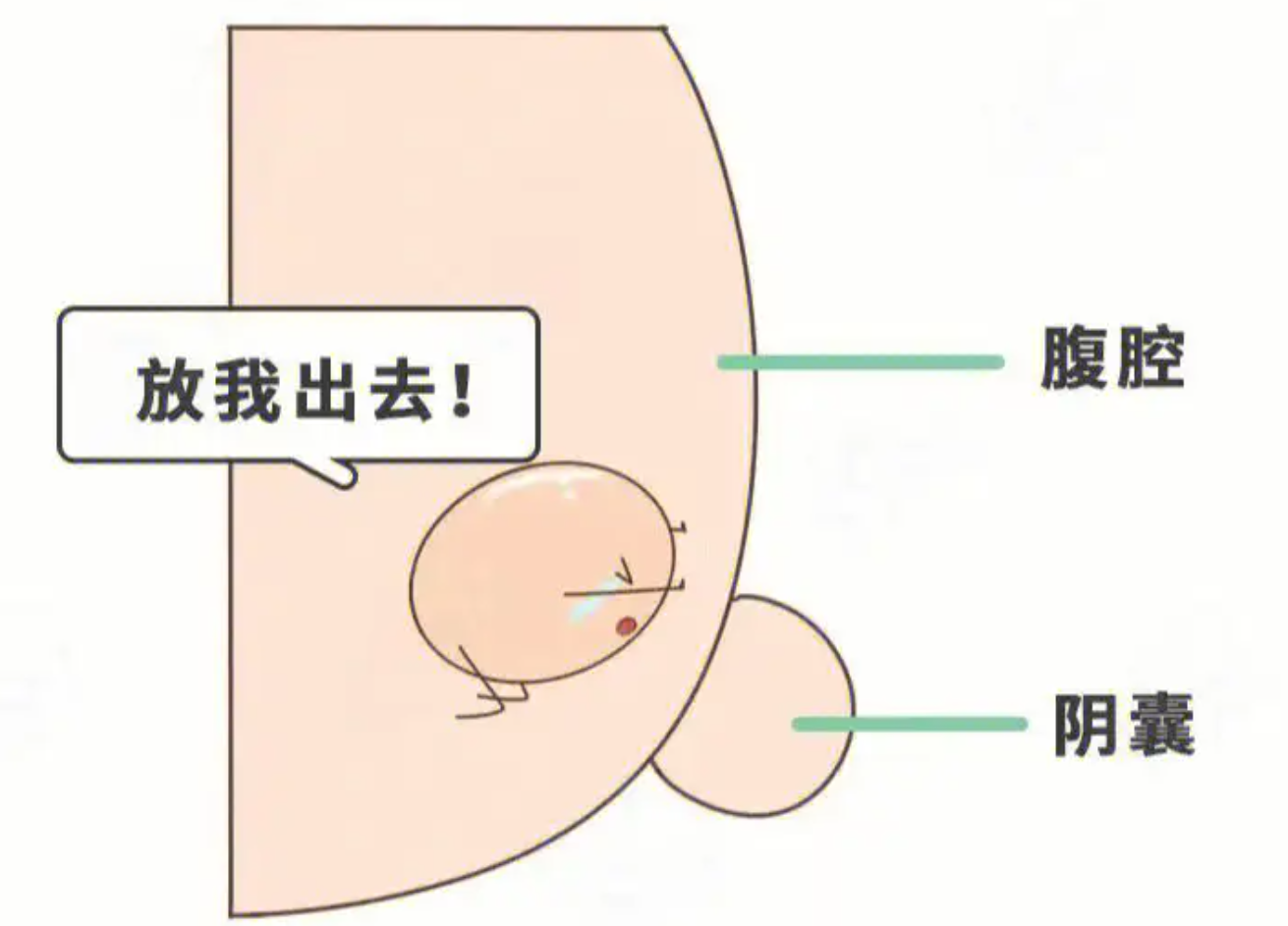 邱志欣 医师 福建医科大学附属第一医院 小儿外科94人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科94人已读 - 多指宝宝怎么办?
多拇指畸形是最常见的先天肢体畸形之一,大多散发,单侧多于双侧,男∶女比例约为2.5∶1。发病率为1/3000~1/1000。我国妇幼卫生监测及年报通讯(2020版)报道2018年多指(趾)发病率为21.40/万,居出生缺陷第2位,但没有提供多拇指畸形的发病率。从患儿父母、甚至非专业医生角度来看,多拇指治疗很简单,“去掉一个,留下一个”即可。如何做好多拇指手术,得到一个外观和功能都满意的结果,比想象的要困难。“六指”其实是多指常见的一种类型,在医学上称为多指畸形,小儿多拇指畸形是小儿外科常见的先天性畸形之一。简单的说,就是除了小儿正常手指外有另外赘生指畸形,常与短指、并指等畸形同时存在。多见于拇指及小指,可单发或多发,但以拇指外侧的单发多指较为常见。这种现象主要是由于遗传以及受环境影响等因素。一、多指畸形分型1.软组织多指:多指仅有软组织赘生物,没有骨、肌腱等组织。2.单纯多指:多指中含有指骨、肌腱和血管神经束与正常手指相连,是一个功能缺陷的手指。3.复合性多指:多指为真正的重复,不仅含有指骨、肌腱等,而且包括掌骨孪生,是完整的外生手指及掌骨。二、出现多指畸形什么时候手术最好呢?需要根据患儿的病情,如发育程度、活动度、畸形严重程度等综合考虑。三、多指畸形如何手术:手术治疗的目的不仅仅是改善外观,更重要的是改进手部的功能,尤其复杂多指畸形需根据具体的形态与功能重建的要求进行关节骨骼畸形矫正、关节囊修补、侧副韧带重建、皮瓣成形等,才能重建一个在功能和外观上最大程度接近正常的手指。
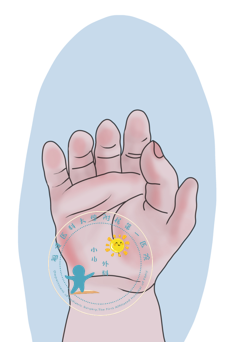 邱志欣 医师 福建医科大学附属第一医院 小儿外科65人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科65人已读 - 妊娠期间B超发现的孩子肾积水怎么办?
先天性肾积水是指在产前及出生后早期,通过超声等筛查发现的肾集合系统扩张。尿液积聚在胎儿肾脏,使肾盂、肾盏扩张,肾实质受压变薄甚至萎缩,进一步可危害肾脏功能。产前胎儿肾积水超声检出率为1%~2%,出生后早期肾积水超声检出率为1%~4%,发病率比较高,值得重视。什么原因导致胎儿先天性肾积水?胎儿先天性肾积水分为生理性和病理性,大多数肾积水为生理性的,病理性肾积水一般由胎儿泌尿系统梗阻导致,发病原因主要有肾盂输尿管连接处狭窄、膀胱输尿管反流等(详见表1)。有些宝妈在产检过程中发现有肾积水,但有时肾积水又消失了。这是什么情况呢?其实这种情况,一般考虑肾积水为生理性的肾积水。一般只需观察随访,无需过多担心。胎儿生理性肾积水的可能原因有:1.孕妇在超声检查前大量饮水,可对胎儿肾脏产生影响,可能通过母体水合作用影响羊水量,间接影响胎儿尿量;2.某些情况下,输尿管近端扩张是由于解剖或功能性梗阻导致的一过性肾积水,在下次检查或出生后逐渐自行消失。3.出生前后,由于肾血管阻力、肾小球滤过率及肾小管浓缩力的不同,使胎儿尿量比出生后增多,造成输尿管、肾盂扩张。怎么发现和鉴别胎儿先天性肾积水?通常在孕妊娠11~14周,胎儿肾脏开始有排尿功能。生理性肾积水主要与胎儿产生尿量多、输尿管收缩功能失调有关,超声表现为肾盂轻度分离,肾实质厚度变化不明显,双侧肾盂扩张程度相当,无明显的输尿管扩张。病理性肾积水根据病因不同、严重程度不同,超声表现也各不相同。有些病理性肾积水超声可明显识别梗阻及部位,而有些需要产后才能做出确切的诊断。怎么评判胎儿先天性肾积水的严重程度?目前常用APD分级系统来评估胎儿肾积水的严重程度(详见表2)。级别越高,提示病情越严重。也有使用SFU分级系统和UTD分级系统进行分级。发现先天性肾积水的胎儿,可以继续妊娠吗?生理性肾积水随着胎儿宫内生长发育进展或胎儿出生后腹腔压力变化,往往可自愈。大多数轻度肾积水病例在分娩前后将自行缓解,而且胎儿或儿童肾积水严重的病例,仍然可能具有一定程度的肾功能。因此,绝大多数肾积水胎儿可发育至足月分娩。但双侧肾重度积水、双侧肾脏发育不良以及进行性的双侧扩张,伴羊水过少和肺发育不良等预后较差,部分胎儿肾积水伴随染色体异常等遗传性疾病。上述情况可由综合性医院相关学科医生组成的会诊团队,根据疾病严重程度、胎龄、孕妇及家属的意愿等具体情况进行综合评估,告知病情后由孕妇和家属决定妊娠的去留。1.宫内治疗目前,先天性肾积水在产前尚无有效的药物治疗,因此,仅能根据疾病严重程度、胎龄、孕妇及家属的意愿等具体情况进行产检随访、胎儿手术干预、早期剖宫产或终止妊娠。 肾积水胎儿期手术干预存在较大争议,目前认为,对于诊断明确且危及胎儿生命的梗阻性疾病所致的肾积水可考虑手术宫内干预,常用的手术方法包括胎儿膀胱-羊膜腔分离术、开放性胎儿手术、胎儿镜手术等,手术时间通常选择在孕20~30周。2.早期剖宫产绝大多数肾积水胎儿可发育至足月分娩,但少数羊水明显减少,且已孕晚期,肺发育成熟患儿可在34周左右行早期剖宫产。3.终止妊娠羊水主要由胎儿尿液形成,严重的羊水过少影响胎儿肺发育,早期胎儿重度肾积水会严重影响胎儿肺和肾脏发育。若超声显示肾脏回声增强、双肾尿路扩张和羊水减少,强烈提示PUV等可能时,可考虑选择性终止妊娠。先天性肾积水胎儿出生后需完善哪些检查?1、胎儿宫内超声诊断肾积水者,应在出生48小时后复查,以避开因暂时的生理性脱水而导致的无尿期。2、对于严重病例(双侧肾积水、孤立肾、羊水过少等)则应出生后立刻行超声检查。3、患儿出生后的超声检查如未发现肾积水也应该于4周后再次复查。先天性肾积水胎儿出生后该怎么处理?先天性肾积水出生后处理主要包括随访与必要的手术干预。规范的随访管理十分重要,生后随访根据风险级别不同,在出生后1~6月内进行超声随访。生理性肾积水往往可自愈。病理性肾积水的治疗包括规律随访和手术治疗。手术治疗指征包括:患侧肾功能受损(GFR<40%),在非手术治疗随访中发现患侧肾功能下降超过10%或超声下肾盂前后径增大,III度、IV度肾积水,合并患侧腰痛、高血压、继发结石形成或者反复尿路感染。
 邱志欣 医师 福建医科大学附属第一医院 小儿外科32人已读
邱志欣 医师 福建医科大学附属第一医院 小儿外科32人已读 - 案例 新生儿空肠闭锁
治疗前男孩,产前2天彩超发现肠管扩张,胎龄38+2周,外院考虑“肠扭转可能”后转诊我科,重新复查超声考虑“空肠闭锁”。治疗中孩子生下来,我们团队完善相关检查后就安排手术,手术前我们通过检查发现孩子的肠管扩张的很大(是正常新生儿的10倍),手术中也证实了我们的观点,并为孩子做了SANTULLI造瘘治疗后治疗后7天手术经过我们团队的精心护理,孩子也有了第一次笑容,孩子妈妈都高兴哭了治疗后3月手术后3个月,孩子体重也长到6kg,达到了关瘘的标准治疗后4月手术切口愈合良好
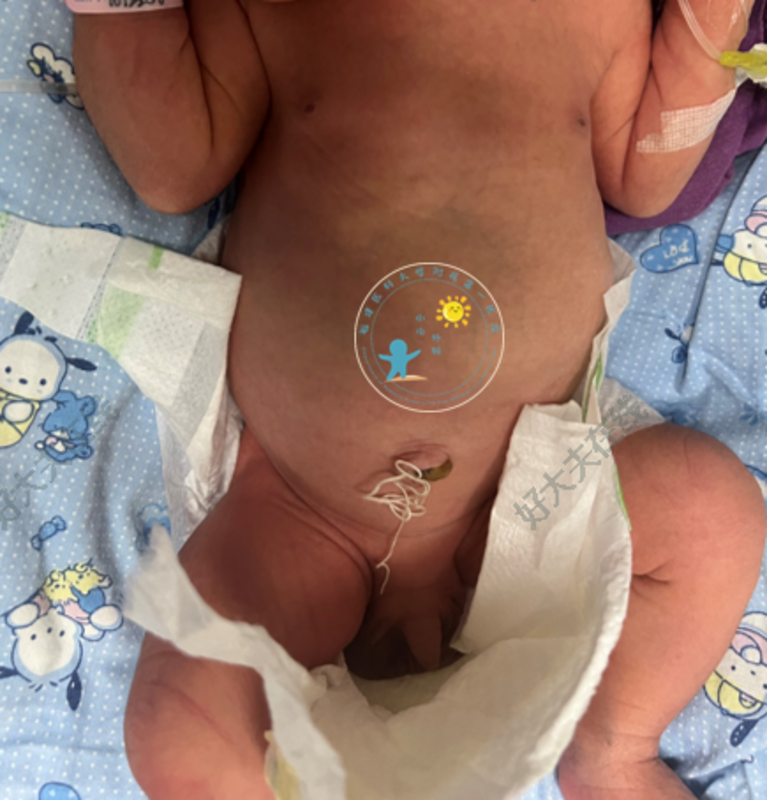 林俊山 主任医师 福建医科大学附属第一医院 小儿外科64人已读
林俊山 主任医师 福建医科大学附属第一医院 小儿外科64人已读

林俊山主任医师
福建医科大学附属第一医院小儿外科
