



赵卫东医生的科普号
- 精选 听力不好,是“听神经瘤”惹的祸?赵卫东,戴春富,黎长江复旦大学眼耳鼻喉科医院耳科中心
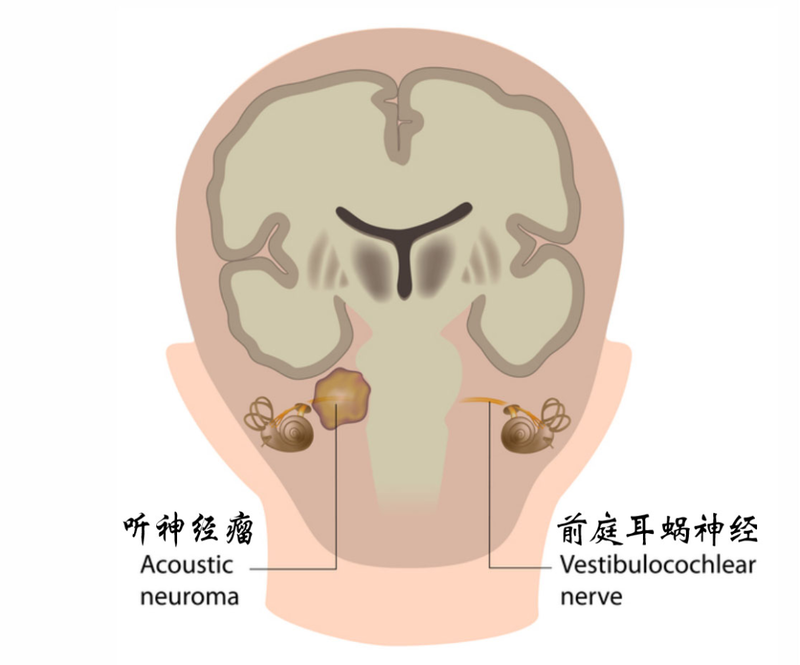
听力不好是一件非常恼人的事情,声音是人们获得外界信息的重要来源之一。这不,前不久就有一位46岁的职业男性,1周前突然出现左耳听力下降,没有耳痛、流水这些症状,却严重影响了他的工作和生活,急忙忙去了当地医院就诊。当地医生按突发性耳聋给予治疗,1个月后仍毫无效果。当地医院就给患者拍了耳部CT,CT结果提示左侧内听道占位性病变。听到“占位”两个字,患者觉得问题严重了,可能耳朵里“长东西”了。 图1 听神经瘤在内听道内 于是为了进一步更好地诊治,来到我们医院,找到专家门诊看诊,给他进一步做了耳部增强MRI的检查,最后结果提示为左侧听神经瘤可能。患者一听是肿瘤,当即紧张万分,乍以为是什么不治之症。专家解释说这是良性肿瘤,是可以治疗的,并不是什么绝症,患者才稍缓焦虑。那既然不是癌症,能否不治,不治疗,会有什么后果?事实上,听神经瘤虽然它暂时不致命,但是如果不治疗,轻则引起听力下降甚至全聋,也可能会压迫面神经导致面瘫,这对容貌和一些面部功能有很大的影响,如果再不处理,任期成长,就可能压迫“脑子”,出现颅内压增高和脑疝,这就可能危及生命了。 图2 听神经瘤可能导致脑疝 那么,“听神经瘤”究竟是一种怎样的疾病?我们该如何看待它? “听神经瘤”这个病名,听起来让人感觉就是长在“听神经”上的肿瘤,这种感觉没错,但却不够精准。之所以会出现这种情况:“全赖最开始给他命名的医生。”医学上其实有很多病名都是这样,开始时不见得认识有那么精确,但因为习惯,所以就会一直延续下去。其实,“听神经瘤”绝大部分都是来源都是“前庭神经”鞘膜的施旺细胞。而真正的能够让人感知到听觉的神经叫“耳蜗神经”,这两种神经常常合在一起并行,难以分离,因而统称为“听神经”。“前庭神经”是用来调节人类平衡的神经,为了理解前庭神经的这一功能,我给大家举个例子。不知道大家有没有这样的生活经验,当你坐在一辆快速行驶的火车上,两旁都是高崖悬壁,而且间距很小,当你用双眼凝视你所对应的悬壁时,你可能会有一种天旋地转的感觉,这种感觉在医学上叫“眩晕”,而它产生的原因,就是“前庭神经”受到激惹所导致的。也就是说“听神经瘤”,更精准的叫法应该是“前庭神经瘤”,极少的人才会发生在“耳蜗神经”上。 图3 看到这幅图所引起的头晕,就是“眩晕” 听神经瘤,是一种很常见的颅内良性肿瘤。它大概占所有脑内肿瘤的8%-10%,如果进一步将脑部细分解剖部位的话,占它所在的桥小脑角区肿瘤的80%-90%,也就是说这一细分部位的肿瘤绝大部分都是它,所以得这个疾病的人,并不“孤独”。30-60岁的人群较其他人群更容易患此病,不过它不挑性别,男女发病率差不多,而且大部分也是只发生在一侧,两侧同时患病或者先后患病的可能性很小。那么,又如何发现和治疗呢?可惜的是,尽管在医学科技如此发达的今天,关于它的发病机制,却依然知之甚少。由于缺乏病因的了解,预防也就无从谈起。我们只能通过尽早发现,尽早治疗这项策略,来减少该疾病对我们的伤害,那么我们如何来早期发现它呢? 若出现听力下降,且渐进性加重,按感音神经性耳聋进行药物治疗又无效时,就要怀疑是这种疾病,因为大部分人是出现这样的情况而被发现的。而至于耳鸣、耳痛、流脓、头晕这些症状都不是必须的,当然也有很少一部分病人是因为这些症状而发现的。但是相比于听力下降,比率还是要少得多。然而,不管怎样,只要出现上述症状,并有加重趋势时,就应该进一步检查,以明确诊断。 图4 听力逐渐下降,一般治疗无效时,应怀疑此病 一旦确诊为“听神经瘤”,该如何治疗?听神经瘤的治疗方案,主要依据肿瘤的大小及目前各脑神经功能状态来决定的。直径小于2.5cm为小肿瘤;直径2.5-4cm为中等大肿瘤;直径大于4cm为大听神经瘤。你就可以根据这个标准,对照CT或MRI的描述来判断自己肿瘤的大小属性了。目前有人主张对于部分较小,又无明显功能损害如无听力下降的患者,建议观察随访。因为有人做了研究,结果显示59%的患者肿瘤年增长速度小于1mm,只有12%的患者年增长速度大于3mm,甚至还有6%的患者肿瘤缩小了。但是随访观察有一个风险,就是听力下降的风险,又有人对此做了研究,小肿瘤和大肿瘤,对听力保存的概率分别是75%和32%,相差一倍。也是说等肿瘤长大了再手术,那么保存听力的可能性就更小。 图5 本案例肿瘤完全切除干净:面神经和听神经也完整保留 因此,对于小肿瘤患者,不介意随着肿瘤增大而听力逐渐下降甚至丧失的,可观察随访;而对保存听力有较为强烈的患者,就算发现时听力无影响,也建议尽早治疗。手术方法有很多。具体选择哪种手术方式,还需结合具体的情况以及医生的习惯来决定。对于一些小肿瘤、不愿手术的患者,也可选择立体定向放疗(伽玛刀),但伽玛刀治疗也有并发症如远期听力丧失等。手术则可以根据具体情况,对听神经和面神经进行很好地保护。如果肿瘤能完全切除,则预后良好。但无论是何种治疗方式,都可能出现并发症,甚至死亡。随着医疗水平的提高,目前手术死亡率小于1%。笔者建议,颅内肿瘤及手术,一旦确诊,还是应尽量寻求经验丰富医生的意见,这样才能改善自己的预后和提高疗效,减少“病急乱投医”。这位患者在我院经手术治疗后,肿瘤完全切除干净,听神经和面神经也得到了完整保护。参考资料:1. 黄选兆,汪吉宝,孔维佳. 实用耳鼻咽喉头颈外科学(第二版)[M]. 北京:人民卫生出版社,2010年.2. 夏寅,张文阳. 听神经瘤治疗策略[J]. 中国耳鼻咽喉颅底外科杂志,2019;25(1):10-14.注:图片选自网络及真实病例。
 赵卫东 副主任医师 上海市五官科医院 颅底神经外科1976人已读
赵卫东 副主任医师 上海市五官科医院 颅底神经外科1976人已读 - 精选 听神经瘤手术方式选择
韩朝 华东医院,赵卫东 复旦大学眼耳鼻喉科医院 听神经瘤的名词其实是一个位置正确、起源错误的诊断,因为后面随着认识的深入,发现听神经瘤绝大多数是来自前庭神经鞘膜的新生物,所以准确的名词是前庭神经鞘膜瘤。但由于听神经瘤绝大多数都在内听道里面(里面主要有耳蜗听神经,前庭神经和面神经经过)生长,所以听神经瘤的诊断还是在日常临床上保留着,患者也好理解。 听神经瘤早期小的时候,会有听力下降、眩晕和耳鸣的症状,因此绝大多数首诊于耳科,但真正在耳科被频繁确诊还是在增强磁共振变的普遍时,也从而引起了治疗方案的改变。磁共振出现之前,CT对于局限于内听道而不破坏骨质的微小听神经瘤的诊出率很低,即使辅助电生理检查ABR也很困难。所以只有长大到出了内听道压迫小脑和脑干才被发现,所以手术多是由脑外科医生通过乙状窦后径路来完成。尽管如此,对于处理内听道底的听神经瘤部分,脑外科医生需要涉及耳科颞骨的处理才能完成。 随着磁共振的发展,局限于内听道的小听神经瘤被频繁发现,而这类听神经瘤由于脑外科手术困难,所以采取两种方式来处理。第一个方式保守观察,如果不是生长很快进而压迫小脑,就不处理;第二个方式是伽玛刀等的放射治疗的兴起,但是随着案例的积累和随访的数据不断增加,发现伽玛刀等放射治疗并不能很好得解决问题,而且增加了手术难度,极少数恶变可能,更不要说保护听力。 基于上述困难,耳神经颅底外科的发展使局限于内听道的微小听神经瘤的切除成为可能,手术基本不进入颅内,是将颞骨上面的硬脑膜抬起,从颅中窝磨除颞骨部分骨质进入内听道,切除听神经瘤。由于耳科医生对颞骨解剖熟悉,手术相对损伤小,安全,从而可以做到保护原有听力,使得听神经瘤手术从单纯的切瘤保命,发展到早期干预保持原有听力的功能保全手术。 所以对于听神经瘤的处理发展到现在根据不同的体积大小和患者年龄有了不同的更加精细的手术方案。 当听神经瘤局限于内听道内,或不超过内听道口1.5cm以内时,可以由耳科医生采用颅中窝入路手术。可能保持原有听力,术后反应很小,属于硬脑膜外操作范畴。 当听神经瘤主要位于内听道内侧涉及压迫脑组织时,需要脑外科医生经乙状窦后入路切除,既可能保护现有听力也可以方便视野清晰的切除肿瘤,缺点是术后反应可能较大。 当听神经瘤位于内听道底向颅内生长但压迫脑组织不严重,并且听力丧失时,可以由耳科医生采用经迷路入路听瘤切除术,这个术后反应小,类似于硬脑膜外操作。这个径路还有一个好处,方便同期人工听觉植入。但对于人工耳蜗植入二期似乎更好。 当患者条件不适合手术或者对手术不能理解时,可以采用伽玛刀控制听瘤生长。
赵卫东 副主任医师 上海市五官科医院 颅底神经外科2928人已读 - 精选 听神经瘤显微外科
听神经瘤起源于内听道里的前庭神经,又称前庭神经鞘瘤,是耳外科最常见的良性肿瘤。其主要症状为缓慢进行性听力下降,直至丧失。肿瘤长大后可突入颅内,造成相应的压迫症状,但肿瘤始终位于脑外,与脑组织之间有明确的界面。听神经瘤的治疗主要有观察和显微手术。观察限于高龄、肿瘤生长极缓慢的患者,而年轻和肿瘤生长较快的患者建议手术治疗。放射外科仅限于无法耐受手术或术后残留者。听神经瘤治疗的现代理念是不仅要求切除肿瘤,更重要的是面、听神经保护和功能重建,这方面耳显微神经外科医师具有独特的优势。耳外科医师能够早期发现肿瘤,并通过对听神经、前庭神经、面神经和脑干功能等进行全面检查和评估,制定最佳治疗方案。耳神经外科医师通常利用耳后硬币大小的骨窗即可切除肿瘤,无需常规开颅,可以不牵拉或者最小程度牵拉脑组织,最大限度减少神经损伤,符合微侵袭和精准治疗的现代理念。术后面、听神经功能重建,更是耳显微神经外科的强项。2015年,上海市听觉医学临床中心引进脑外科医师。耳神经外科与脑外科的融合,是一个国际趋势。融合脑外科的路径和方法,采用乙状窦后径路和颅中窝径路,可以在保留面神经功能的同时,最大可能的保留听力。复旦大学眼耳鼻喉医院耳显微外科、耳神经外科在王正敏院士开拓性的工作和卓越的领导下,在迟放鲁教授、李华伟教授、陈兵教授和戴春富教授等领衔的治疗团队的共同努力下,在听神经瘤诊疗方面积累了丰富经验。2015年起开创了听神经瘤手术新模式。本文系赵卫东医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
赵卫东 副主任医师 上海市五官科医院 颅底神经外科5792人已读 - 医学科普 听神经纤维瘤病(NF2) 孙皓洁 任冬冬 袁雅生 赵卫东
青少年皮肤突然出现多个咖啡牛奶斑或粉色软瘤?单侧或双侧听力下降或耳鸣?警惕听神经纤维瘤病(NF2)!01什么是听神经纤维瘤病?听神经纤维瘤病(NF2)也叫Ⅱ型神经纤维瘤病,是由于NF2基因突变导致的常染色体显性遗传肿瘤综合征,一种因内听道前庭神经鞘膜细胞异常增殖导致的良性肿瘤,最常临床表现为双侧的听神经瘤,双侧发病占所有NF2相关听神经瘤的85%,常伴发神经系统肿瘤及眼和皮肤等相关病变。02什么年龄最容易得听神经纤维瘤病?NF2的发病年龄大多在20~60岁,大部分病人在20岁左右出现相关症状,部分患者会伴发脑膜瘤、室管膜瘤、其他部位的神经鞘瘤(如脊髓)等其他肿瘤,青少年和儿童时期也可发病,患病率约为1/60000。虽然临床上大部分患者发现患病时都在20岁左右,但是并不意味着年长的人就可以完全排除这个疾病。NF2相关听神经瘤主要可分为两个亚型:(1)Wishart型:症状较为严重,多在20岁前发病,疾病进展迅速,最终导致双侧听力丧失,常合并有颅内或椎管内肿瘤。(2)Gardner型:轻型,发病较晚,疾病进展缓慢,合并颅内或椎管内肿瘤较少。03为什么我会得听神经纤维瘤病?NF2相关听神经瘤是一种常染色体显性遗传病,由22号染色体上的NF2基因变异引起,约一半的该病患者有明确的家族遗传,也有患者是新发突变。NF2基因突变的比例和形式不同导致该疾病的患者临床症状和疾病进展情况各不相同,遗传给后代的风险也因人而异。图:NF2基因突变引起神经鞘膜细胞异常增生04听神经纤维瘤病有什么症状?NF2的主要症状包括听力下降(最初通常为单侧)、耳鸣和眩晕(走路不稳、头晕)。恶心呕吐、严重的眩晕(天旋地转感)在疾病后期才会出现。但是这些症状并不具有特异性,许多其他疾病也可以有同样的临床表现,所以要及时来医院就诊进行科学的检查。除了听神经的症状以外,也有一些患者因为其他部位肿瘤的症状就医发现:如脑膜瘤(头痛、癫痫发作)、脊髓肿瘤(肌肉无力、肢体感觉异常)等;眼部症状也常见,通常表现为弱视、斜视和白内障;皮肤肿瘤、斑块(咖啡牛奶斑)、皮下肿瘤也可能是NF2患者的临床表现,占到NF2患者的70%。如果听神经瘤持续生长压迫面神经和三叉神经,可能会出现面瘫的症状。体积过于巨大的肿瘤可能会压迫脑组织,严重的可以威胁生命。图:NF2患者的皮肤表现05如果我怀疑自己或家人有听神经纤维瘤病,应该怎么办?当出现听力下降、耳鸣或眩晕时,应及时前往耳鼻喉科或神经外科就诊,进行听力学和前庭功能检查,然后进行颅脑及脊柱的CT和MRI检查明确诊断。建议患者同时进行基因检测,明确发病原因。图:听神经瘤NF2增强MRI和CT的表现,红色圆圈标记双侧桥小脑脚听神经肿瘤图:耳鸣和眩晕是Ⅱ型神经纤维瘤病的常见症状06听神经纤维瘤病如何治疗?对于病情进展迅速的病人,如不进行手术切除,可能会威胁生命,手术是治疗NF2最主要的方法,手术分为保留和不保留听力两种方式,当肿瘤超过2cm时,听力基本不能保留。如双侧肿瘤较小、生长缓慢,可以随访观察,每年进行MRI和听力检查。不能耐受手术的患者可以采用立体定向放射治疗控制肿瘤生长,提高生活质量,目前尚无有循证医学有效的药物治疗,多数处于研究阶段。目前手术仍然是治疗NF2相关听神经瘤的首要选择。NF2病人术后的听力康复有两种选择:人工耳蜗植入(CI)和听觉脑干植入(ABI),其中人工耳蜗植入目前被认为效果更好,前提是手术后耳蜗神经仍然完整,因此需要结合每个患者的不同情况制定治疗方案。图:人工耳蜗植入及人工脑干植入示意图07如果已经诊断听神经纤维瘤病,需要注意什么?虽然NF2是一种良性肿瘤,但往往会导致严重的后果,比如双侧听力丧失,失明,反复多次手术切除颅内和脊柱占位,引起重要神经功能的丧失,最严重的是威胁生命。因此对于这部分患者,建议尽早学习手语和唇语,及早发现和治疗,听力保留和重建治疗是NF2治疗的核心,另外,由于这种疾病是遗传疾病,诊断明确的患者在考虑生育前建议进行产前咨询,并在孕期进行胎儿基因诊断,减少遗传给子代的风险。# 参考文献 #CoyS,RashidR,Stemmer-RachamimovA,SantagataS.AnupdateontheCNSmanifestationsofneurofibromatosistype2[publishedcorrectionappearsinActaNeuropathol.2019Aug20. ActaNeuropathol.2020;139(4):643-665.doi:10.1007/s00401-019-02029-5HallidayJ,RutherfordSA,McCabeMG,EvansDG.Anupdateonthediagnosisandtreatmentofvestibularschwannoma.ExpertRevNeurother.2018;18(1):29-39.doi:10.1080/14737175.2018.1399795EvansDG.Neurofibromatosistype2(NF2):aclinicalandmolecularreview.OrphanetJRareDis.2009;4:16.Published2009Jun19.doi:10.1186/1750-1172-4-16
 赵卫东 副主任医师 上海市五官科医院 颅底神经外科1419人已读
赵卫东 副主任医师 上海市五官科医院 颅底神经外科1419人已读

赵卫东副主任医师
上海市五官科医院颅底神经外科
