



上海市奉贤区皮肤病防治所皮肤科科普号
- 上海某区老年特应性皮炎临床特征分析_
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科110人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科110人已读 - Mal de Meleda病: G86R突变一例(一种罕见的掌跖角化病)
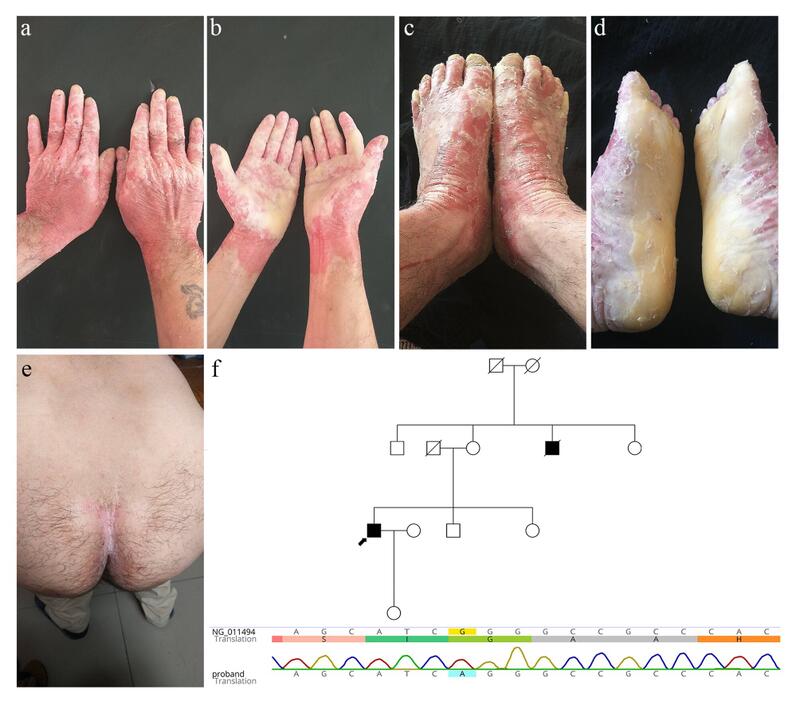
MaldeMeleda病:G86R突变一例祝英1,张丹露1,郭韫懿1,郭碧蓉2,孙忠辉1(1.上海奉贤区皮肤病防治所皮肤科,上海201408;2.安徽医科大学第三附属医院皮肤科,安徽合肥230000)[摘要]MaldeMeleda(MDM)属于一种罕见的掌跖角化病,是一种常染色体隐性遗传疾病,由SLURP1基因突变引起。G86R是热点突变。到目前为止,已经鉴定出至少20个患者含此突变在许多国家报道,包括台湾、巴勒斯坦、土耳其、爪哇、巴基斯坦、利比亚和韩国,中国大陆也有二家系报道。我们报告一MaldeMeleda病例:中年男性含G86R突变且并发甲真菌病,目的是丰富MDM样本数据库,加深对该病的认识。[关键词]基因突变;MaldeMeleda病;甲真菌病AG86RmutationmaldeMeledapatientwithonychomycosisZHUYing,ZHANGDan-lu,GUOYun-yi,GUOBi-rong,SUNZhong-hui(DepartmentofDermatology,FengxianInstituteofDermatosisPreventionandTreatment,Shanghai201408,China)[Abstract]MaldeMeleda(MDM)belongstoarareformofPPK,whichisautosomalrecessiveandcausedbymutationsintheSLURP1gene,withpossiblefoundereffects.Sofar,thereareatleast20mutationsthathavebeenidentified.G86Risahotspotmutationandiscurrentlyreportedinmanycountries,includingTaiwan,Palestine,Turkey,Java,Pakistan,Libya,Korea,andChina.Although,twoChinesemainlandreportsofithavebeenpublishedinrecentliteratures.Wereportamiddle-agedmalecasewithonychomycosis,withtheaimofenrichingtheMDMsampledatabasetodeepentheunderstandingofthedisease.[Keywords]genemutation;MaldeMeledadisease;onychomycosis基金项目:皮肤病学教育部重点实验室(安徽医科大学)开放课题基金资助项目(AY2017-1-017);上海市奉贤区科委基金资助项目(20161119和20171003)通讯作者:孙忠辉,E-mail:szhgyy3344@163.com或郭碧蓉,E-mail:guobr1983@163.comMaldeMeleda病(MDM)属于一种罕见的掌跖角化病(PPK),该病是常染色体隐性遗传,由基因SLURP-1突变引起。到目前为止,已经鉴定出SLURP-1基因有至少20个致病突变。G86R是热点突变,目前在许多国家报道,包括台湾、巴勒斯坦、土耳其、爪哇、巴基斯坦、利比亚、韩国和中国,中国大陆目前已有两个家系被报道。我们报告一例中年男性含G86R突变且并发甲真菌病的患者,目的是丰富MDM样本数据库,以增加对该病的了解。1病历摘要先证者,男性,47岁,主诉:掌跖角化增厚、伴有脱屑和多汗45年。出生后,先证者的手和足逐渐出现皮肤增厚和弥漫性红斑。经常有脱屑、出汗和异味。在过去的10年里,皮肤增厚和红斑逐渐增多,常伴有瘙痒。此外,指、趾甲增厚变形、发黄。皮肤科检查:手部皮肤红斑持续到腕关节上约5cm,呈手套状;足部红斑持续到踝关节以上约5cm,呈袜套状。手足背、掌基底都为弥漫性红斑。手足掌部为弥漫性的蜡黄色角化斑块。手背和足背脱屑,部分区域有渗出,呈湿疹样改变。指趾甲变形、增厚,尤其是脚趾趾甲。尾骨区域沿臀沟有浸润性红斑(图1a、b、c、d、e)。牙齿、头发和智力均正常,各系统检查未见明显异常。先证者否认家族遗传史。父母非近亲结婚,但是来自河南省的同一个村庄,而且一个死去的叔叔患有类似疾病。(图1f)先证者基因检测发现SLURP-1中有一个纯合错义突变c.256G>A(p.G86R)(图1f)。手足的角化斑块,氢氧化钾(KOH)直接显微镜检查,检测到真菌阳性(正常为阴性)。左拇趾甲真菌培养7天后,生长为白色菌落、背面呈黄褐色,可见到螺旋菌丝和棒状大分生孢子(×200倍)(图2)。该真菌被鉴定为须癣毛癣菌。诊断:MaldeMeleda病治疗:由于先证者有HBsAg(+),他拒绝口服抗真菌药和阿维A胶囊,接受了局部治疗,主要为酮康唑软膏和丙酸氯倍他索乳膏。因此,患者皮损没有明显改善。2讨论MDM是一种罕见的常染色体隐性遗传性掌跖角化病。在1826年由LucaStulli在克罗地亚Meleda岛发现并在意大利杂志首次报道了该病,其患病率约为1/10万[1]。临床体征主要为双侧弥漫的掌跖角化,颜色为蜡黄色、呈袜状和手套状,边缘清晰。患者在出生后不久开始出现掌跖红斑,并进展为特征性的可剥脱的角化斑块。最终,过度角化也可以出现在手和脚的背部表面(越线现象transgrediens),transgrediens会从儿童时期的手指背部开始,到成年时逐渐发展到手和脚的背部。过度角化可并发多汗症,随后的微生物感染会导致恶臭和皮损疼痛[2]。指甲异常也是最常见的特征,包括甲下角化过度、Beau’s线(甲横沟症),甲真菌病、厚甲、反甲。患者也可能有手指的功能损害,包括第五指发育不良,手指变细,挛缩,关节垫和假指断症[3]。银屑病样皮损也可存在于膝部和肘部。也可伴有口周红斑和唇炎。在疾病的后期,在MaldeMeleda的角化过度区域内可出现恶性黑色素瘤[4]。Meleda角化病需要与长岛型掌跖角化病(NPPK)、Greither型进行性掌趾角化病、Bothnia型非表皮松解型掌跖角化病等鉴别。本研究小组曾报道一例NPPK[5],症状相对温和,皮损虽累及手足背部、腕内侧、踝和跟腱部位。但角化斑块不显著,红斑也较本先证者轻。在2001年,Fischer等确认MaldeMeleda病与编码SLURP-1蛋白的基因突变有关[6]。SLURP-1的参与调节角质形成细胞生长、增殖、分化和凋亡。存在于表皮的颗粒层,广泛分布于人体皮肤、宫颈、牙龈、胃和食管,在掌跖含量最高。蛋白失活时,角质形成细胞凋亡不能被正常调控,导致皮肤高度角化[7]。该蛋白抑制巨噬细胞和角质形成细胞释放肿瘤坏死因子。失活时会引起持续炎症,最后导致肿瘤[8]。迄今为止,全世界不同人群中已检测到至少20个SLURP-1基因的致病突变位点[9]。既往早期研究显示,MDM除在亚得里亚海的Meleda岛有较高发病率外,中东和地中海地区也有。在这些地区近亲结婚较为常见,提示祖先效应。截至2018年4月,目前人类基因突变数据库(HGMD)和人类基因变异数据库(LOVD)显示已有包括阿尔及利亚、土耳其、巴勒斯坦、巴基斯坦、中国台湾、德国、苏格兰、日本、利比亚等多达10余个国家的病例报道。本研究先证者根据临床表现和基因诊断结果为MDM。SLURP-1最常见的突变是C28fs32X,W15R和G86R[9]。G86R目前在六个种族中得到发现,包括中国人[10,11],印度尼西亚人[9],韩国人[12],利比亚人[1],巴勒斯坦人[13]和巴基斯坦人[14]。2016年我国Zhang[10]等首次报道两个通过基因检测确诊为MDM的家系,3例儿童患者均发生c.256G>A(G86R)错义突变。即SLURP-1第3外显子中第256位核苷酸G→A发生纯合的无义突变。G86R在中国大陆人中Pan[11]还报道了一名中年妇女,她的特点是手指屈曲挛缩。在台湾,Chao[15]和Tjiu[4]分别报道了了具有假显性遗传的女性患者家系和一例T细胞活化缺陷的老年患者。作为中国大陆的第三例报道,与上述文献中的患者相比,我们的先证者是一名中年男性并明确鉴定患有甲真菌病。由于掌趾皮肤的过度角化、增厚和出汗,掌跖角化病患者手足通常容易感染真菌。然而,上述G86R文献中的那些中国患者的临床症状并未显示真菌感染;国外G86R突变的其他人群中也没有关于甲真菌病的报道。但是国内有学者报道了对称性进行性红斑角化病并发真菌感染的三例患者。作者猜想可能与皮肤中某些蛋白成分或功能异常和相关基因位点的突变有关[16]。我们注意到这一点,但是本例患者是否的确与G86R突变造成的真菌易感性有关,尚需大样本的临床研究探讨其具体原因。本文只是想强调随着年龄的增长,MDM患者的病情会越来越严重,一些患者甚至会发展患有肿瘤。同样,MDM患者也可能会受到长期真菌感染的影响,它将严重影响患者的生活质量,应及早干预。参考文献[1]BchetniaM,BozgiaM,LaroussiN,etal.ThefirstMaldeMeledacaseinLibya:identificationofaSLURP1mutation[J].2015,54(12):1426-1428.[2]MoraiseSilvaFA,CunhaTV,BoenoEdosS,etal.MaldeMeleda:areportoftwocasesoffamilialoccurrence[J].AnBrasDermatol,2011,86(4Suppl1):S100-103.[3]MarrakchiZ,MarrachiS,MeziouTJ,etal.MaldeMeleda.16cases[J].TunisMed,2006,84(7):423-426.[4]TjiuJW,LinPJ,WuWH,etal.SLURP1mutation-impairedT-cellactivationinafamilywithmaldeMeleda[J].2011,164(1):47-53.[5]孙郅劼,郭韫懿,张丹露,等.长岛型掌跖角化病:一例纯合缺失的SERPINB7基因突变[J].皮肤病与性病,2019,41(02):157-159.[6]FischerJ,BouadjarB,HeiligR,etal.MutationsinthegeneencodingSLURP-1inMaldeMeleda[J].HumMolGenet,2001,10(8):875-880.[7]FavreB,PlantardL,AeschbachL,etal.SLURP1IsaLateMarkerofEpidermalDifferentiationandIsAbsentinMaldeMeleda[J].JInvestDermatol,2007,127(2):301-308.[8]ChimientiF,HoggRC,PlantardL,etal.IdentificationofSLURP-1asanepidermalneuromodulatorexplainstheclinicalphenotypeofMaldeMeleda[J].HumMolGenet,2003,12(22):3017-3024.[9]RadionoS,PramonoZAD,OhGGK,etal.IdentificationofnovelhomozygousSLURP1mutationinaJavanesefamilywithMaldeMeleda[J].2017,56(11):1161-1168.[10]ZhangJ,ChengR,NiC,etal.FirstMaldeMeledareportinChineseMainland:twofamilieswitharecurrenthomozygousmissensemutationinSLURP-1[J].2016,30(5):871-873.[11]PanY,ZhaoH,ChenA,etal.AMalDeMeledapatientwithsevereflexioncontracturesofhandsandfeet:AcasereportinWestChina[J].2017,96(36):e7972.[12]OhYJ,LeeHE,KoJY,etal.ASporadicCaseofMaldeMeledaCausedbyGeneMutationinSLURP-1inKorea[J].2011,23(3):396-399.[13]EcklKM,StevensHP,LestringantGG,etal.MaldeMeleda(MDM)causedbymutationsinthegeneforSLURP-1inpatientsfromGermany,Turkey,Palestine,andtheUnitedArabEmirates[J].2003,112(1):50-56.[14]WajidM,KurbanM,ShimomuraY,etal.MutationsintheSLURP-1geneunderlieMaldeMeledainthreePakistanifamilies[J].2009,56(1):27-32.[15]ChaoSC,HuangCY,LaiFJ,etal.PseudodominantinheritancewiththeG86RmutationintheARSgeneinMaldeMeleda[J].2006,45(12):1456-1458.[16]吴凡,陈军,任芳,等.对称性进行性红斑角化病并发真菌感染3例[J],临床皮肤科杂志2014,43(12):732-734.图示:Legendstofigures:图1:先证者主要临床表现(a、b、c、d、e),家系图及基因诊断结果(f),参考序列:NG011494。Figure1:Clinicalfeaturesoftheproband(a,b,c,d,e),Pedigreeandgeneticresults(f),referencesequence:NG011494.图2:左拇趾甲真菌培养(a)及鉴定(b、c)(×200倍)。Figure2:Fungalculture(a)andidentificationoftheleftthumbtoenail,(b,c)(200X).
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科242人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科242人已读 - 长岛型掌跖角化病:一例纯合缺失的SERPINB7基因突变
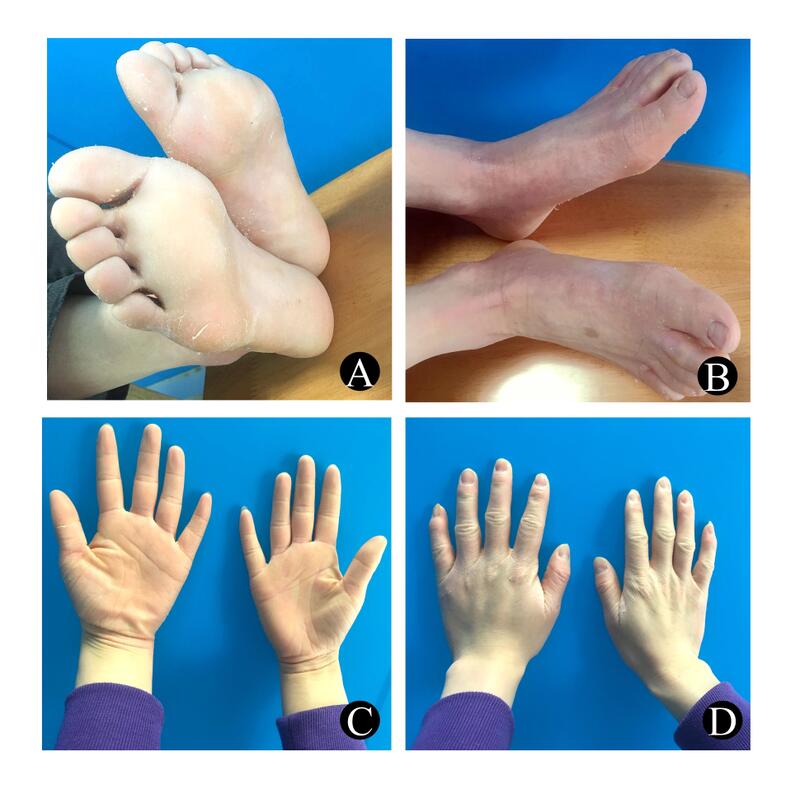
长岛型掌跖角化病:一例纯合缺失的SERPINB7基因突变孙郅劼1,郭韫懿1,张丹露1,祝英1郭碧蓉2,孙忠辉1[摘要]:目的报道1例长岛型掌跖角化病,确定其致病基因突变。方法在先证者家系调查的基础上,收集家系患者和正常人的血样,并采集正常对照血样100份,采取聚合酶链反应技术对长岛型掌跖角化病致病基因SERPINB7基因进行扩增,并对其产物进行测序。结果先证者存在SERPINB7基因7号外显子的c.650-653delCTGT(p.S217Lfs7)纯合突变。先证者父母为杂合缺失。结论SERPINB7基因的c.650-653delCTGT(p.S217Lfs7)纯合突变是引起患者长岛型掌跖角化病的原因,这是该疾病、该位点作为纯和突变首次报道。[关键词]:长岛型掌跖角化病;基因;SERPINB7:突变Nagashima-typePalmoplantarKeratoderma:acaseofhomozygousdeletionoftheSERPINB7genemutationSUNZhijie1,GUOYunyi1,ZHANGDanlu1,ZHUYing1,GUOBirong2,SUNZhonghui1(1.DepartmentofDermatology,FengxianInstituteofDermatosisPreventionandTreatment,Shanghai201408,China;2.DepartmentofDermatology,TheThirdAffiliatedHospitalofAnhuiMedicalUniversityandTheFirstPeople’sHospitalofHefei,AnhuiHefei,230061China)[Correspondingauthors]:SUNZhonghui,Email:szhgyy3344@163.com;GUOBirong,Email:guobr1983@163.com[Abstract]:ObjectiveToreportacaseofNagashima-typepalmoplantarkeratoderma(NPPK),andtoidentifydisease-causemutationsintheSERPINB7gene.MethodsBasedontheinvestigationofthefamilyoftheproband,thebloodsamplesofthefamilyandnormalpeoplewerecollected,and100normalbloodsampleswerecollected.PCRwasperformedtoamplify8exonsandtheirflankingsequencesoftheSERPINB7genefollowedbyDNAsequencing.ResultsTheprobandhadahomozygousmutationintheSERPINB7genec.650-653delCTGT(p.S217Lfs7).Theproband‘sparentswereheterozygousConclusionThehomozygousmutationofc.650-653delCTGT(p.S217Lfs7)ofSERPINB7geneisthecauseofNPPKinproband.Thiscaseisthefirstreportashomozygousc.650-653delCTGT(p.S217Lfs7).ofSERPINB7genemutation.[Keywords]:Nagashima-typepalmoplantarkeratosis;Gene;SERPINB7;Mutation[基金项目]:皮肤病学教育部重点实验室(安徽医科大学)开放课题基金资助项目(AY2017-1-017);上海市奉贤区科委基金项目(20171003)[作者单位]:1.上海市奉贤区皮肤病防治所皮肤科,上海201408;2.安徽医科大学第三附属医院暨安徽省合肥市第一人民医院皮肤科皮肤科,安徽合肥230061[通讯作者]:孙忠辉,E-mail:szhgyy3344@163.com;郭碧蓉,E-mail:guobr1983@163.com长岛型掌跖角化病(Nagashima-typepalmoplantarkeratosis,NPPK,OMIM615598)是一种常染色体隐性遗传病,表现为掌趾部位红斑和角化斑块、脱屑,伴手足多汗;皮损会影响手背、足背、腕部、踝部、跟腱等部位。NPPK属于弥漫性掌跖角化病。掌跖角化病(palmoplantarkeratoderma,PPK)是以掌跖皮肤过度角化为主要特点的一组复杂疾病。目前已鉴定的25种遗传性PPK病可分为五类[1]:(1)弥漫性PPK;(2)弥漫性残毁性PPK;(3)局灶性PPK;(4)伴PPK的外胚层发育不良;(5)综合征性PPK。Kubo等[2]于2013年将NPPK的致病基因确定为编码丝氨酸蛋白酶抑制因子B亚型7的SERPINB7基因。本研究报道一NPPK家系,检测结果显示先证者具有纯合突变c.650-653delCTGT(p.S217Lfs7)。这在既往文献未见显示,现将结果报道如下。1资料与方法1.1临床资料先证者,女,29岁,出生后半年掌跖部位即出现弥漫性红斑,累及手、足背部,腕部及足踝和跟腱,伴手足多汗。近十年手足出现轻度异味,遇水后发白。皮肤科检查:先证者双手掌趾弥漫性角化增厚,表面呈淡黄色;手足背、指趾背侧面、手腕内侧、足踝区和跟腱区有红斑、脱屑;指、趾甲正常。见图1。父母为近亲结婚,但手足未见皮损。否认家族有类似患者。1.2、外周血DNA提取:获得知情同意书后,抽取患者及其家属外周血2ml。应用QIAampDNAbloodminikit(QIAGEN公司,德国)提取基因组DNA,标化质量浓度至10ng/µ。另外,以同样的方法提取100例无亲缘关系的健康对照个体基因组DNA作为对照。1.3、PCR扩增和直接测序:通过NCBI(http://www.ncbi.nlm.nih.gov/)查取SERPINB7基因序列,采用Primer5.0软件设计设计特异性引物对NF1基因的编码外显子2-8进行PCR扩增。扩增产物纯化后直接在ABIPRISM®3700测序仪上测序(AppliedBiosystems),测序结果与人类基因组SERPINB7基因序列(NG_034150)比较。使用Chromas2.2软件进行解读,并用Geneious11.0软件进行比对分析。2结果将所测得的序列与SERPINB7基因序列(NG_034150)比较,得到如下结果:c.650-653delCTGT(p.S217Lfs7)丢失4个碱基CTGT,出现移码突变。即第7外显子CDS第650处丢失4个碱基CTGT,导致移码突变。原来的编码的蛋白质第217处TCT丝氨酸变为TTA亮氨酸,至第7个密码子移码为终止密码子TGA,出现终止。父母均为杂合突变。所有突变均由反向测序得到验证。同时在100例无亲缘关系的正常人中进行此基因直接测序,未发现上述突变,见图2。3讨论NPPK是常染色体隐性遗传病。1977年,日本学者Nagashima等首次报道该病,由此而命名[3]。NPPK最初被认为是MaldeMeleda掌跖角化病一种较轻的临床亚型。2013年,Kubo等对3个家系的日本NPPK患者进行全外显子组测序,确定SERPINB7基因为NPPK的致病基因。SERPINB7基因位于18q21.3,共有8个外显子,第一个不编码。编码丝氨酸蛋白酶抑制因子B亚型7,具有抑制丝氨酸蛋白酶的作用。SERPINB7功能的缺失可能导致NPPK皮损处蛋白酶活性过强。SERPINB7主要表达于表皮颗粒层及角质层上部[2]。目前,SERPINB7基因共发现了12种突变,包括无义2,移码7,剪接点2和错义突变1[2,4-10]。SERPINB7有两处活性位点[11]。由螺旋C和螺旋D构成的CD-loop,分别位于46–62和74–89位,即基因位置的3号外显子编码处。目前有p.41_42del[4]和p.Q73Lfs17[2]可能影响该活性。另外一处活性位点RSL(Reactivesiteloop)位于第331~352位氨基酸环。剩余的10个已知突变所致蛋白截断位置都介于两活性位点之间,所以都影响后者活性。活性位点环不能表达,将会造成患者表皮颗粒层和角质层SERPINB7蛋白酶活性抑制作用丧失,丝氨酸蛋白酶活性不能有效抑制,从而导致角质形成细胞蛋白降解,角质层屏障功能破坏。因而患者手足易多汗,皮肤容易遇水肿胀发白。根据形态学,NPPK属于遗传性弥漫性掌跖角化病。通常表现为出生时至3岁以内出现掌跖部位弥漫性界清红斑,伴有轻度至中度角化;常累及手足背部,腕内侧、踝和跟腱也是好发部位,肘膝部也可受累。遇水后角化皮肤可出现发白改变。皮疹稳定,不随年龄增长出现明显进展或加重。患者常伴有手掌和足底多汗,易伴发皮肤浅表真菌感染。组织病理无特异性,患者一般情况良好,无毛发、甲、牙齿等其他外胚层受累表现,也无其他器官系统受累表现。弥漫性掌跖角化病中,症状具有多汗、遇水易肿胀发白的主要有NPPK、MaldeMeleda掌跖角化病、Bothnia型掌跖角化病[12]、进行性掌跖角化病(Greither病)等鉴别。见表1。迄今为止,文献显示SERPINB7基因突变存在建立者突变c.796C>T(p.R266)(rs142859678),该突变的杂合在日本和中国的正常人群中携带频率分别为2/89和6/197[13]。目前国内文献报道的患者突变位点也是与这有关[13-15]。另外,已经报道的NPPK病例,都是为日本、中国和韩国学者[16]发现。因而可以推测目前该病主要存在于东亚人群。本文所发现的突变首先由北京大学杨勇教授等报道[8]。但报道的患者为复合杂合突变,另一突变位点为日本、中国最常见的建立者突变c.796C>T(p.R266)。该文献中先证者父母已亡故,无法验证。本研究所报道的父母是携带同一杂合突变,而且是存在近亲关系,为一典型常染色体隐性遗传模式家系。本文所发现的纯合突变丰富了SERPINB7基因突变数据库。虽然是首次报道,但在先证者临床表型与其他已报道患者没有大的区别。推测双c.650-653delCTGT也是影响了活性位点RSL的功能。目前NPPK逐渐被国内外学者认识,国际公认的诊断标准还没建立。由于存在建立者突变,因此在国内应该还有许多患者没有被诊断明确。同时,掌跖角化病致病基因众多,这会更迫使我们在诊治此类患者时需要结合患者病史,临床表现和实验室检查并通过基因检测最终来明确NPPK诊断。参考文献[1]PerezC,KhachemouneA.MaldeMeleda:AFocusedReview[J].AmJClinDermatol.2016,17(1):63-70.[2]KuboA,ShiohamaA,SasakiT,etal.MutationsinSERPINB7,encodingamemberoftheserineproteaseinhibitorsuperfamily,causeNagashima-typepalmoplantarkeratosis[J].AmJHumGenet.2013,93(5):945-956.[3]NAGASHIMAM.Palmoplantarkeratoses[M]//MIURA,OCHIAIK.HandbookofHumanGenetics.Tokyo:IgakuShoin,1977:23-27..[4]ZhangJ,ZhangG,NiC,etal.Nagashima-typepalmoplantarkeratosisinaChineseHanpopulation[J].MolMedRep.2016,14(5):4049-4054.[5]NakajimaK,IshiguroM,ShiohamaA,etal.Novelframe-shiftmutationinSERPINB7inaJapanesepatientwithNagashima-typepalmoplantarkeratosis[J].JDermatol.2017,44(7):841-843.[6]KatsunoM,ShiohamaA,AokiS,etal.NovelnonsensemutationinSERPINB7andthetreatmentoffootodorinapatientwithNagashima-typepalmoplantarkeratosis[J].JDermatol.2017Jul;44(7):e146-e147.[7]MizunoO,NomuraT,SuzukiS,etal.HighlyprevalentSERPINB7foundermutationcausespseudodominantinheritancepatterninNagashima-typepalmoplantarkeratosis[J].BrJDermatol.2014,171(4):847-853.[8]YinJ,XuG,WangH,etal.NewandrecurrentSERPINB7mutationsinsevenChinesepatientswithNagashima-typepalmoplantarkeratosis[J].JInvestDermatol.2014,134(8):2269-2272.[9]ShiohamaA,SasakiT,SatoS,etal.IdentificationandCharacterizationofaRecessiveMissenseMutationp.P277LinSERPINB7inNagashima-TypePalmoplantarKeratosis[J].JInvestDermatol.2016,136(1):325-328.[10]HuaS,MiaoX,MaoW,etal.AnovelframeshiftSERPINB7mutationinaChinesecasewithNagashima-typepalmoplantarkeratosis:casereportandreviewoftheliterature[J].ClinExpDermatol.2018Jun11.doi:10.1111/ced.13682.[Epubaheadofprint][11]SuzukiS,NomuraT,MizunoO,etal.IdentificationofpreviouslyunknownSERPINB7splicevariantsinpatientswithNagashima-typepalmoplantarkeratosisrevealstheimportanceoftheCD-loopofSERPINB7[J].BrJDermatol.2015,173(5):1288-1290.[12]CaoX,YinJ,WangH,etal.MutationinAQP5,encodingaquaporin5,causespalmoplantarkeratodermaBothniatype[J].JInvestDermatol,2014,134(1):284-287.[13]戴珊,南栩,赵红珊,等.长岛型掌跖角化病:SERPINB7基因突变位点研究[J].中国中西医结合皮肤性病学杂志,2017,(02):108-112.[14]刘成,李常兴.长岛型掌跖角化病一家系丝氨酸蛋白酶抑制剂B7基因的突变分析[J].皮肤性病诊疗学杂志,2018,(01):8-11.[15]多丽娜,汪慧君,林志淼,等.长岛型掌跖角化病二例SERPINB7基因突变研究[J].中华皮肤科杂志,2016,(3):180-182.[16]OnHR,LeeSE,NomuraT,etal.IdentificationofSERPINB7mutationsinKoreanpatientswithNagashima-typepalmoplantarkeratosis[J].JDermatol.2017,44(7):840-841.附图:图1.先证者手足皮损先证者双手掌趾弥漫性角化增厚,角质层呈淡黄色。手足背、指趾背侧面、手腕内侧、足踝和跟腱有红斑、脱屑。指甲及趾甲正常。图2先证者及其父母SERPINB7基因直接测序结果参考序列:NG_034150。先证者:纯合c.650-653delCTGT(p.S217Lfs7),父母均为杂合突变。附表:表1四型掌跖角化伴多汗的鉴别[2]GreitherBothnianMaldeMeledaNagashimaOMIM144200600231248300615598遗传模式常染色体显性遗传常染色体显性遗传常染色体隐性遗传常染色体隐性遗传致病基因KRT1AQP5SLURP1SERPINB7患病率罕见罕见1/100,000日本:1.2/10,000中国:3.1/10,000发病年龄8-10月童年幼年出生一年内病理非表皮松解非表皮松解非表皮松解非表皮松解角化过度厚轻至厚严重轻手、足背皮损++++多汗++++遇水变白−+−+皮损影响它处肘部、膝盖、弯曲区域和跟腱−膝盖和肘部,腔口周和眶周红斑肘部、膝盖和跟腱自发性截肢+−偶尔−屈曲挛缩−−+−
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科250人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科250人已读 - 表皮松解性掌跖角化病二家系基因突变检测
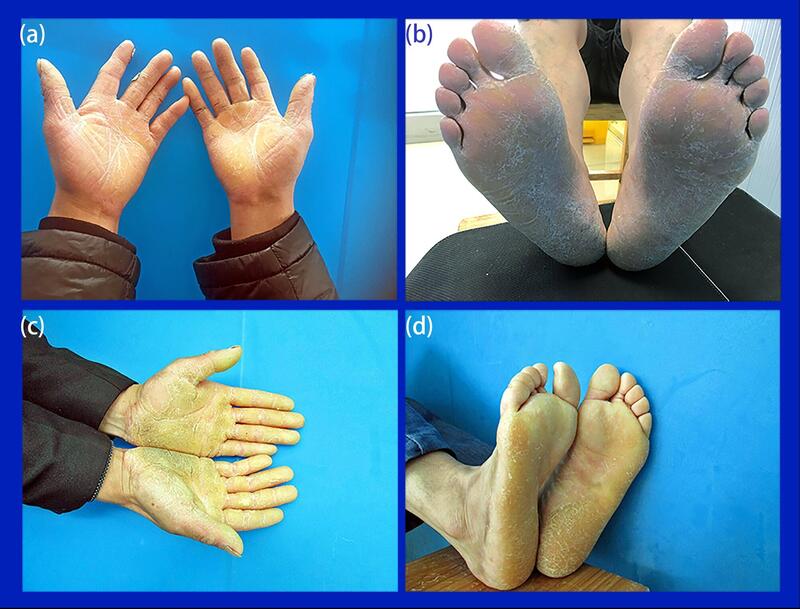
表皮松解性掌跖角化病二家系基因突变检测郭韫懿、孙忠辉[摘要]目的:研究表皮松解性掌跖角化病家系患者的临床表现及其致病原因。方法:在二表皮松解性掌跖角化病家系家系调查的基础上,收集二家系4例患者和其家属的血样,同时采集(无亲缘关系)正常对照血样100份,采取聚合酶链反应技术对KRT1、KRT9和KRT16基因进行扩增,并对其产物进行测序。结果:发现家系1先证者的KRT1基因含杂合突变c.598T>C(p.F200L)。发现家系2先证者及子女的KRT9基因含杂合突变c.488G>A(p.R163Q)。而家系正常成员及家系外无亲缘关系的100个正常对照中均不存在此突变。结论:二个发生的突变均为已报道过的重复突变,其中KRT1基因突变p.F200L是国内首次报道。该二表皮松解性掌跖角化病家系家系发病原因是KRT1、KRT9基因发生突变所致。研究结果进一步证实:一些单基因遗传性疾病存在遗传和临床异质性。[关键词]表皮松解性掌跖角化症;异质性;基因突变KRT1andKRT9mutationsresultinepidermolyticpalmoplantarkeratoderma:reportoftwoChinesefamiliesGUOYunyi1,ZHUYing1,ZHANGDanlu1,GUOBirong2,SUNZhonghui11.DepartmentofDermatology,FengxianInstituteofDermatosisPreventionandTreatment,Shanghai201408,China;2.DepartmentofDermatology,TheThirdAffiliatedHospitalofAnhuiMedicalUniversity,TheFirstPeople’sHospitalofHefei,Hefei230022,ChinaCorrespondingauthor:SUNZhonghui,E-mail:zhgyy3344@163.comorGUOBirong,E-mail:guobr1983@163.comObjective:Tostudytheclinicalmanifestationsandcausesofetiologyoffamilyepidermolyticpalmoplantarkeratoderma(EPPK).Methods:BasedonthefamilysurveyofEPPK,wecollectedbloodsamplesfromprobandsandrelativesoftwoEPPKfamilies,and100unrelatednormalcontrolbloodsamples.Then,theKRT1,KRT9andKRT16genesweresequencedbydirectsequencing.Results:ItwasfoundthattheKRT1geneinthefamily1proband1washeterozygousforc.598T>C,p.F200L,andproband2offamily2andhischildrencontainedmutationsc.488G>A,p.R163QinKRT9gene.Thismutationwasinvisibleinthenormalcontrolsofthisfamilyaswellastheadditional100unrelatednormalcontrols.Conclusion:Thetwomutationshavebeenreportedasrecurrentmutations,ofwhichtheKRT1genemutationp.F200LwasfirstreportedinChina.ThecauseofthepedigreesofourEPPKfamilieswerethemutationsofKRT1andKRT9genes.WeshouldpayattentiontothegeneticandclinicalheterogeneityinthediagnosisandtreatmentofEPPKinclinicalpractice.[Keywords]epidermolyticpalmoplantarkeratoderma;heterogeneity;genemutation基金项目:皮肤病学教育部重点实验室(安徽医科大学)开放课题基金资助项目(AY2017-1-017);上海市奉贤区科委基金资助项目(20161119和20171003)作者单位:1上海市奉贤区皮肤病防治所皮肤科,上海,2014082安徽医科大学第三附属医院,安徽省合肥市第一人民医院皮肤科皮肤科,合肥,230022通信作者:孙忠辉,E-mail:szhgyy3344@163.com或郭碧蓉,E-mail:guobr1983@163.com表皮松解性掌跖角化病(diffuseepidermolyticPPK,EPPK)主要临床表现是手掌和足趾发生过度角化,是一种常染色体显性遗传病[1]。目前研究显示该病是由不同致病基因引起:KRT1、KRT9和KRTl6,因此该病具有遗传异质性。本课题组2017年诊断两个表皮松解性掌跖角化症家系,分别为KRT1、KRT9基因突变。其中KRT1基因突变p.F200L是国内首次报道。本文根据我们的基因诊断结果结合文献讨论EPPK的异质性,现报道如下。材料和方法1、临床资料1.1先证者1,男性,58岁,工人。因双掌跖角化50余年,于2015年8月来我院就诊。先证者自4岁起无明显诱因双掌跖部出现对称性片状红斑,角化,形状不规则,受累皮肤粗糙增厚,一般无自觉症状,有时伴有瘙痒、疼痛。皮疹逐渐向周围扩展,至15岁时已累及双手整个掌侧及双足跖,部分皮疹成蜡黄色角化性斑块;指(趾)甲未见增厚、混浊。23岁后皮疹未再扩展。期间双掌、双足部皮疹反复发作,有一定季节性,冬重夏轻,伴有足部多汗(图1,a、b)。家族史:调查家族中4代人。先证者父母非近亲结婚,外祖父、母亲(均已亡)有此病史,先证者儿子正常。符合常染色体显性遗传模式(图2,a)。体格检查:一般情况良好,各系统检查未见明显异常。皮肤科检查:皮疹分布于双手掌、双足底部,主要表现为境界清楚的黄色角化斑块、丘疹,伴脱屑。1.2先证者2,男性,37岁,工人。因双掌跖角化30余年,于2015年6月来我院就诊。先证者自5岁起无明显诱因双掌跖部出现对称性片状红斑,角化,形状不规则,受累皮肤粗糙增厚,通常无自觉症状,有时伴有瘙痒、疼痛。皮疹逐渐向周围扩展,至20岁时已累及双手整个掌侧和双足跖,部分皮疹成坚硬蜡黄色角化斑块;指(趾)甲未见变形。20岁后皮疹未再扩展。期间双掌,双足部皮疹反复发作,呈角化过度,脱落,再角化,交替进行,有一定季节性,冬重夏轻。(图1,c、d)。家族史:调查家族中3代人。先证者父母非近亲结婚,父亲发病(已亡);目前家族中共有患者3人,其中男性2人,女性1人。先证者儿子、女儿发病,符合常染色体显性遗传模式(图2,b)。体格检查:一般情况良好,各系统检查未见明显异常。皮肤科检查:皮疹分布于双手掌、双足底部,主要表现为境界清楚的黄色角化斑块、有皲裂。两例先证者病理相似:角化亢进,表皮增生,粒层增厚,棘层肥厚,真皮浅层血管周围稀疏淋巴组织细胞漫润。颗粒层及棘层未见裂隙及颗粒性空泡变性,未见表皮松解(图2,e、f)。2、外周血DNA提取:获得知情并签署同意书后,2家系患者及亲属抽取外周血3ml。其中,家系1患者仅为先证者1;家系2患者为先证者2和一子、一女。应用QIAampDNAbloodminikit(QIAGEN公司,德国)提取基因组DNA,标化质量浓度至10mg/µ。另外,以同样的方法提取100例无亲缘关系的健康个体基因组DNA作为对照。3、PCR扩增及DNA测序:通过NCBI(http://www.ncbi.nlm.nih.gov/)查取KRT1、KRT9和KRT16基因核苷酸序列,Primer3.0设计KRT1、KRT9和KRT16外显子特异性引物(htttp://www.genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi)。PCR扩增后,所有PCR产物经纯化后直接在ABIPRISM®3700测序仪上测序(AppliedBiosystems)。测序结果与人类基因组KRT1、KRT9和KRT16基因核苷酸序列比较。使用Chromas2.2软件进行解读,并用Geneious11.1进行比对分析。结果PCR产物经测序发现:患者1家系先证者KRT1基因CDS第598处杂合T碱基成为C碱基,因此编码的苯丙氨酸变为亮氨酸。即:c.598T>C,p.F200L。经检索HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)、LOVD(http://www.lovd.nl/3.0/home)数据库该突变是国内首先报到(图2,a)。家系2先证者及子女KRT9基因CDS第488处杂合T碱基成为A碱基,因此编码的精氨酸变为天冬酰胺。即:c.G488>A,p.R163Q。该突变为该病常见突变(图2,b)。而两个家系的家系内正常对照及家系外100个无亲缘关系的正常人中均不存在此突变。2图1两先证者临床症状和病理。(a),(b)先证者1;(c),(d)先证者2。两先证者手掌和足底都有角化斑块。(e),(f)为先证者1病理,HE染色,(e)×40,(f)×200。图2家系图和测序结果。(a)家系1,参考序列(NG_008364);(b)家系2,参考序列(NG_008300)。(标“↑”者为先证者;标“+”者为行基因突变检测)讨论掌跖角化症(palmoplantarkeratoderma,PPK)是一组掌跖角化疾病的统称,存在二十几种亚型。根据临床症状发生形态的不同,PPK可以分为3种类型:弥漫性(diffuse),局灶性(focal)和点状性(punctate)。由于是仅根据临床形态来分型,而每一型往往致病基因有多个,所以总的来说PPK是遗传异质性很强的一组疾病。表皮松解性掌跖角化症(EPPK)是弥漫性掌跖角化症的一个亚型,OMIM号为144200。根据国际人类中间丝数据库(HumanIntermediateFilamentDatabase,HIFD,http://www.interfil.org/)截止2014年11月22日的数据显示,目前明确的致病基因有KRT1、KRT9和KRT16。我们在基因诊断EPPK时,虽将KRT16基因列入侯测基因,但至今未发现致病突变。既往报道过一例EPPK是KRT16突变引起,也仅是罕见的嵌合型[2],目前的文献表明所有KRT16基因突变基本都与先天性厚甲症[3]有关。因此EPPK,主要致病基因是KRT1和KRT。EPPK属于常染色体显性遗传,多为婴儿期开始发病,轻者仅有掌跖皮肤粗糙,重者掌跖处出现表皮松解性斑块状、边缘清晰的角质增厚,呈黄色胼胝体样。有时角质增厚可蔓延至掌跖皮肤侧缘或手足背,可伴有指节垫、甲板增厚浑浊、指趾屈曲畸形等。病理:表皮高度角化过度,颗粒层及棘层增厚,颗粒层及棘层;可有表皮松解,即可见裂隙及颗粒性空泡变性。EPPK与非表皮松解性掌跖角化病(NEPPK)本质是同一病,因临床改变是一样的,且都是KRT1、KRT9突变引起。NEPPK又名“Thost”型掌跖角化症,早在1880年由Thost发现并命名。2002年Kuster等重新检测了Thost患者的后裔,结果组织病理还是显示有表皮松解[4]。本研究致病突变检测上,家系1先证者的KRT1基因突变p.F200L是2016年Gagliardi[5]已报道;而家系2的KRT9基因突变p.R163Q是目前国内外均已报道的热点突变。虽然两先证者组织病理都未见表皮松解现象,但结合临床症状和基因诊断结果,两家系患者都诊断为EPPK。KRT9角蛋白局限性的表达于掌跖表皮中[6],突变引起的疾病仅是EPPK。KRT9基因虽然异质性不强,但存在突变热点区和热点突变。数据库显示KRT9基因突变热点区位于1A区前端的157-172个氨基酸位置。在总共25个突变中位于1A区域有20个,位于2B区仅5个。基因突变频率最高的是第163氨基酸突变,Liu[7]统计该位置约占所有报道病例的44%,而p.R163W约占29.33%,其次为p.R163Q约占10%。第163位氨基酸突变可能影响角蛋白中间丝组装,从而导致EPPK。本研究先证者2家系正是携带此突变,因而是一明确的EPPK.相较于KRT9角蛋白,KRT1角蛋白主要表达在全身表皮的棘层和颗粒层细胞,其突变可以引起众多表型。目前显示除了EPPK,表皮松解性角化过度型鱼鳞病(EHK)、先天性大疱性鱼鳞病样红皮病(BCIE)、周期性鱼鳞病伴表皮松解性角化过度(CIEH),豪猪状鱼鳞病(IHCM)、弥漫性非表皮松解性掌跖角化症(NEPPK)、条纹状掌跖角化症(SPPK)、常染色体显性遗传脑白质营养不良(ADLD)和Greither综合征(GS)也与其突变有关。HIFD数据库显示KRT1突变目前有47个,大多数(32个)是引起表型相对严重的BCIE,而表型相对较轻的PPK(包括EPPK,NEPPK)仅有8个,均为错义突变。引起BCIE的突变绝大多数位于角蛋白1A和2B区域。KRT1中1A和2B是编码体内角蛋白丝装配的重要区域,该区域改变会影响角蛋白丝的形成并影响其稳定性,进而影响角蛋白网状结构形成[8]。在8个引起PPK的突变中,影响的编码序列分别位于头(Head)、1A、1B和2B区域,未显示突变热点。PPK有遗传异质性,同样在KRT1基因也表现出了复杂的临床异质性。KRT1同一突变可在家系内引起不同表型,比如Nellen[9]描述一家系携带c.1016T>A,4岁先证者是严重的表皮松解EHK。她不仅有EPPK和屈曲部位的疣状增厚,还有躯干部位过度角化伴和去除鳞屑后的糜烂。而患者家系其他患者仅有EPPK和皮肤摩擦后出现水泡。同样的,KRT1同一突变可在家系以外引起不同表型。比如国内学者报道的一例EPPK[10]与国外报道[11]的BCIE和CIEH的致病突变竟然都是KRT1错义突变c.1436T>C。然而,本研究先证者1的突变p.P200L目前文献显示仅一例为意大利报道[5]。Gagliardi报道采用外显子测序发现一家系中既有Charcot-Marie-Toothdisease(进行性神经性肌萎缩综合征,CMT)又有PPK的患者4例,其他患者单独患有CMT或PPK各1例。作者认为这两个疾病是单独引起,致病突变分别是MPZ基因的p.Ser44Phe和KRTI基因p.P200L。p.P200L位于1A区域,经蛋白预测软件SIFTPred、Polyphen2和MutationTaster都认为是致病突变。由于其合并其它基因突变、表型复杂,p.P200L所致EPPK的病情是否发展、有无异质性,还需随访观察。我们报道的也是同一突变PPK家系,符合文献,但无其它合并基因突变疾病。由于KRT1基因突变存在上文所述的复杂的临床异质性,所以先证者1还是需要随访、以观察其病情发展。另外,本突变在中国人群是首例报道。综上所述,本研究的结果及文献复习提示我们在临床实践中对于诊断和治疗一些单遗传疾病要注意其遗传和临床异质性。参考文献[1]王唯嘉,康晓静.表皮松解性掌跖角化病研究进展[J].国际皮肤性病学杂志,2016,(2):95-98.[2]TerrinoniA,PudduP,DidonaB,etal.AmutationintheV1domainofK16isresponsibleforunilateralpalmoplantarverrucousnevus[J].JInvestDermatol,2000,114(6):1136-40.[3]WilsonNJ,O‘TooleEA,MilstoneLM,etal.Themoleculargeneticanalysisoftheexpandingpachyonychiacongenitacasecollection[J].BrJDermatol,2014,171(2):343-55.[4]KüsterW,ReisA,HenniesHC.EpidermolyticpalmoplantarkeratodermaofVörner:re-evaluationofVörner‘soriginalfamilyandidentificationofanovelkeratin9mutation[J].ArchDermatolRes,2002,294(6):268-72.[5]GagliardiS,RiccaI,FerrariniA,etal.PalmoplantarkeratodermaandCharcot-Marie-Toothdisease:combinationoftwoindependentgeneticdiseases?IdentificationoftwopointmutationsintheMPZandKRT1genesbywhole-exomesequencing[J].BrJDermatol,2017,177(1):284-286.[6]LangbeinL,HeidHW,MollI,etal.Molecularcharacterizationofthebodysite-specifichumanepidermalcytokeratin9:cDNAcloning,aminoacidsequence,andtissuespecificityofgeneexpression[J].Differentiation,1994,55(2):164.[7]LiuWT,KeHP,ZhaoY,etal.ThemostcommonmutationofKRT9,c.C487T(p.R163W),inepidermolyticpalmoplantarkeratodermaintwolargeChinesepedigrees[J].AnatRec(Hoboken),2012,295(4):604-9.[8]ArinMJ,OjiV,EmmertS,etal.Expandingthekeratinmutationdatabase:novelandrecurrentmutationsandgenotype-phenotypecorrelationsin28patientswithepidermolyticichthyosis[J].BrJDermatol,2011,164(2):442-7.[9]NellenRG,NagtzaamIF,HoogeboomAJ,etal.Phenotypicvariationinepidermolyticichthyosis:clinicalandfunctionalevaluationofthenovelp.(Met339Lys)mutationintheL12domainofKRT1[J].ExpDermatol,2015,24(11):883-5.[10]钱佳丽,臧东杰,周城,等.一个表皮松解性掌跖角化病家系的KRT1基因突变分析[J].中华皮肤科杂志,2011,(4):232-234.[11]AustinSmithW,CopeA,FernandezM,etal.Infantileepidermolyticichthyosiswithprominentmaternalpalmoplantarkeratoderma[J].DermatolOnlineJ,2016,22(4).
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科216人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科216人已读 - 引用 先天性鱼鳞病致病基因研究进展
DOI:10.3760/cma.j.issn.1673。4173.2013.05.014作者单位:201408上海奉贤区皮肤病防治所皮肤科(孙忠辉、郭韫懿);上海交通大学医学院附属新华医院皮肤科(李明、姚志荣)通信作者:姚志荣,Email:zryaosmu@sohu.con本文主要缩写:Iv:寻常性鱼鳞病,FLG:中间丝聚合蛋白,EHK:表皮松解性角化过度型鱼鳞病,IB:Siemens大疱性鱼鳞病,ARCI:常染色体隐性遗传鱼鳞病,LI:板层状鱼鳞病,NCIE:非大疱先天性鱼鳞病样红皮病,HI:丑角样鱼鳞病,STS:类固醇硫酸酯孙忠辉,郭韫懿李明姚志荣【摘要】鱼鳞病是由许多疾病组成,其中很大一部分是遗传性疾病。这种疾病通常是表皮最先被影响,其主要特征是在临床上表现为全身皮肤干燥、粗糙和鱼鳞状鳞屑,组织学上绝大部分是角质层增厚。先天性鱼鳞病至少有20种,随着遗传技术的进步,此类疾病已经得到了不少认识。该病的发生与组成皮肤屏障的基因突变有关。这些基因编码:角质细胞形成有关的结构蛋白,细胞连接蛋白,细胞连接水解、脂质代谢及DNA修复所需酶类。此类疾病由于遗传异质性和临床异质性原因分类非常复杂。根据遗传模式进行分类,可更好了解基因型-表型之间的关系。本文根据遗传模式不同,概述这一组疾病的致病基因研究进展。【关键词】先天性鱼鳞病;遗传模式;基因;突变AdvancesincongenitalichthyosispathogenesisgeneresearchSUNZhong-hui,GUOYun-yi,LIMing,YAOZhi-rong.DepartmentofDermatology,FengxianInstituteofDermotosisPrevention,Shanghai201408,ChinaCorrespondingauthor:YAOZhi-rong,Email:zryaosmu@sohu.com【Abstract】Ichthyosisiscausedbyanabnormalityinskingrowththatresultsindryingandscaling.Itcanoccurasadiseaselimitedtotheskinorinassociationwithabnormalitiesofotherorgansystems.Histologically,thevastmajoritycharacterizethickeningofthestratumcorneum.Theichthyosesareagroupofskindiseases,ofwhichlargepartsareinheritedforms.Thereareatleast20typesofcongenitalichthyosis.Geneticapproachestounderstandingcongenitalichthyosishaverevealedthegenedefectsunderlyingmanyofthesegenodermatoses.Theoccurrenceisrelatedtothegenemutationwiththecompositionoftheskinbarrier.Thesegenesencode:keratinocytesrelevantstructuralprotein,thecelljunctionprotein,cellconnectionhydrolysisrequiredforlipidmetabolismandDNArepairenzymes.However,classificationisverycomplexduetogeneticheterogeneity.Classificationbasedongeneticpatterncanbetterunderstandtherelationshipbetweengenotype-phenotype.Thisreviewmainlyprovidesanoverviewoftheprogressofthepathogenicgeneticstudiesinthosecongenitalforms,accordingtothedifferentgeneticpatterns.【Keywords】congenitalichthyosis;geneticpattern;gene;mutation鱼鳞病是由许多疾病组成,其中很大一部分是遗传性疾病。这种疾病通常是表皮最先被影响,其主要特征是在临床上表现为全身皮肤干燥、粗糙和鱼鳞状鳞屑,组织学上绝大部分是角质层增厚。先天性鱼鳞病是由于先天遗传缺陷引起的一组疾病。此类疾病由于遗传异质性和临床异质性原因分类非常复杂。本文根据遗传模式不同,对这一组疾病的致病基因研究进展做一概述。常染色体半显性遗传寻常性鱼鳞病(IV)是泛发性鱼鳞病中最常见,也是症状最轻的。患者生后数月出现白色或灰色鳞屑对称分布于背部及四肢伸侧,尤其双下肢。掌跖可有过度角化及线纹增多,毛周角化也很常见。可并发特应性皮炎、哮喘,症状冬重夏轻,多数患者青春期后病情减轻。研究发现中间丝聚合蛋白(FLG)与IV有关。中间丝聚合蛋白的编码基因是FLG,该基因位于1号染色体,长度约为23.03kb,含有3个外显子,外显子3长度为12573bp,其编码序列为12048bp,它是由10-12重复序列(972bp)组成。外显子1序列非编码,外显子2包含起始密码子,而几乎整个中间丝聚蛋白原都由外显子3编码。中间丝聚蛋白在维持表皮屏障的通透性、调节皮肤正常pH值、防御微生物对皮肤的侵犯等都起重要作用。McKinley-Grant等(1989)首先克隆了cDNA。Smith等(2006)证实FLG突变与IV有关,两个突变R501X和2282del4均位于FLG重复序列1中。他们认为IV属于半显性遗传病,R501X纯合子表现为中度患者,R501X/2282del4杂合子具有严重的IV表现。Palmer等(2006)研究显示FLG的R501X和2282del4突变在欧洲人中与特应性皮炎(AD)和哮喘有关。在欧洲虽然FLG有新突变报道但最普遍的仍是R501X和2282del4。Nomura等(2007)[1]对7例日本IV患者FLG测序发现突变S2554X和3321delA,在143例AD患者也发现了此二种突变。无论是患者还是正常对照都未发现R501X和2282del4。ZhangH等(2010)[2]在对261例中国汉族AD患者研究发现3321delA和K4671X是高频突变。以上表明FLG高频突变在IV和AD人种上有不同分布。FLG突变与银屑病易感也有关。Hu等(2012)[3]发现在一IV与银屑病并存的家系中有K4022X。此突变纯和仅发现在银屑病患者,而杂合却在IV和正常人发现。值得注意的是Liu等(2008)[4]在对两个IV家系连锁分析研究时,确认了新的位点,定位在染色体10q22.3-q24.2。常染色体显性遗传表皮松解性角化过度(EHK),又称先天性大疱性鱼鳞病样红皮病(BCIE),为常染色体显性遗传。临床表现为出生短时间内突发泛发性红斑大水泡鳞屑,数月内消失。约第三,四年起皱褶区及伸侧出现角化过度性线状疣样皮损,掌跖呈板样角化。以后发展为广泛的角化过度,持续终生。病理表现为典型的棘层松解角化过度。电镜下角质形成细胞核周的中间丝聚集成团块,这些聚集团块被K1和K10特异的抗体所标记。KRT1、KRT10基因是EHK的致病基因。Compton等(1992)用连锁分析将EHK基因定位染色体12q11-q13的KRT1基因。Lessin等(1988)则定位于17q12-q21的KRT10基因。此后关于KRT1、KRT10基因突变的检测多有报道,以点突变为主,螺旋起始区及螺旋末尾区是突变热点。EHK的限局型:表皮痣(Epidermalnevi)是沿Blaschko线分布局部增厚的色素沉着过度皮损。虽然在临床很难区分,但EHK表皮痣病理上呈典型表皮松解角化过度。父母如具有此种表型,后代往往具有泛发性EHK。这种嵌合型突变KRT1、KRT10基因突变发生在受精卵形成之后的胚系发育阶段,患者外周血及正常皮肤中均检测不到,只有在皮损处的表皮细胞才可检出。Siemens大疱性鱼鳞病(IBs)由KRT2基因突变引起的,与EHK的表型极为相像。出生时即有广泛水疱,继而为角化过度性鳞屑所替代,主要累及四肢屈侧皮肤,不伴红皮病。电镜显示患者棘层和颗粒层角质形成细胞内中间丝聚集,颗粒层细胞间有松解性角化过度。该病也为常染色体显性遗。Steijlen等(1994)用连锁分析定位于12染色体编码2型角蛋白的区域。KRT2热点突变区位于KRT2的杆状区高度保守的羧基末端,但也有在H1区[5]。IBs因与EHK表型相似,当无KRT1、KRT10基因突变时需检测KRT2基因。Bygum等(2012)[6]在对来源12个家系的16位EHK患者临床和突变检测中发现:5个家系是KRT1突变,6个家系是KRT10突变。1个家系尽管临床诊断为IBs,也未检测出KRT1和KRT10突变,但仅查到了位于外显子1上缺失18个碱基的多态。该研究发现EHK中自发突变为75%远高于既往50%,在螺旋区的突变表型较严重,而且KRT1突变患者都有掌跖角化症。EHK中,其它涉及角蛋白基因突变的常染色体显性遗传疾病。高起鱼鳞病致病基因为KRT1,环状表皮松解性鱼鳞病致病基因为KRT1、KRT10。总的来说,EHK本身是一个病名。但在组织病理上,是一组编码角蛋白的基因突变所引起的单基因疾病的共同特征。又如豪猪状鱼鳞病实际上是症状学命名。目前分Brocq型,Lambert型,Bafverstedt型,Curth—Macklin型,Rheydt型。Brocq型病理上也是表皮松解性角化过度。常染色体隐性遗传常染色体隐性遗传鱼鳞病(ARCI)。主要是指板层状鱼鳞病(LI)和非大疱先天性鱼鳞病样红皮病(NCIE)。最初LI就是指所有ARCI,上世纪八十年代ARCI逐渐分为LI和NCIE。但在临床上仍有许多病例,无论在组织病理或是电镜超微结构都无法将两者严格区分。临床上90%此二类患者为火棉胶婴儿,但LI在早期一个月内会死于败血症和蛋白电解质紊乱或有的病人会彻底痊愈,也有病人会长期具有类似寻常鱼鳞病表型。NCIE相较于LI具有红皮病是特征性的,病人伴有掌跖角化过度、手指挛缩、半数病例伴有甲损害包括甲粗糙不平、指甲下疣状增生或角化不全,病变还可累及到头皮,睫毛和眉毛脱落的发生率高于板层状鱼鳞病;但是眼睑外翻、口唇外翻却较LI发生率低。因此Akiyama[]认为此二者仍是同一种角化疾病,是一组异质性很强的疾病。但承认目前将二者划分是有益于治疗的。ARCI的发病机制可能与表皮最终分化过程中角质细胞套膜(CE)蛋白异肽键形成异常或与表皮脂质屏障的形成障碍有关。涉及的致病基因主要有:TGM1、ALOXE3、ALOX12B、ABCA12。TGM1基因,位于染色体14,TGMl基因编码表皮转谷氨酰胺酶酶—1是表皮表达的三种TGM的主要亚型,也是与膜相连的钙依赖性硫醇酶,可催化角质蛋白CE前体蛋白间ε一(γ谷氨酰)赖氨酸交联的形成,提高膜结构的稳定性,防止水分缺失和细菌感染。Yamanishi等(1992)使用原位荧光杂交技术将TGMl确认在在14q11.2。Farasat等(2009)[8]认为相较错义突变,其中至少有一种截断突变与LI出生时即有的少汗过热严重症状有关。编码精氨酸的密码子有高频突变,主要是CpG二核苷酸的脱氨引起。最常见突变是在内含子5的A→G换位,占总突变的28%。ARCI临床症状与TGM1基因突变呈明显相关的有:出生时火棉胶、睑外翻、碟形鳞屑、脱发。ALOXE3和ALOXA12B基因分别编码脂加氧酶-3和12-R脂加氧酶。与速发型过敏和炎症有关的白三烯可由花生四烯酸经脂加氧酶催化而制得。脂加氧酶-3和12-R脂加氧酶是表皮型脂加氧酶,可通过此途径参与皮肤的终末分化,调节皮肤屏障的发育、形成、功能及影响其外观与结构。NCIE皮肤的渗透屏障作用是失常的,此二酶加速经皮肤水分缺失和表皮细胞有丝分裂,因此可出现鱼鳞病症状。Eckl等(2005)研究17个NCIE家系它们11个点突变在ALOXE3基因,p.Arg234X和p.Pro630Leu为热点突变;5个点突变在ALOXA12B基因。2009年又在对250名ARCI患者研究发现此二基因是除TGM1以外的第二致病基因,以上二突变依然是热点突变[9]。ABCA12基因,编码ATP结合盒转运子A12蛋白,是ATP结合盒式转运蛋白超家族成员。ABCA12是角质包膜中参与脂质转运的角质细胞脂质转运子,它的功能缺失导致角质层脂质转运出现障碍。免疫电镜显示位于人类表皮上层的角质形成细胞。Lefevre等(2003)研究LI,发现4个错义突变在ATP结合域,此区域为高度保守区。Nawaz等[10]发现了一新突变p.G1559V,尽管也发生在此区域但症状较轻。ABCA12基因也是ARCI中丑角样鱼鳞病(HI)致病基因。它是鱼鳞病中表型最严重、最易发生死亡的疾病。患儿在出生时有外貌奇异,坚硬的盔甲包被体表使面部变形呼吸和喂养受限。可有显著的睑外翻唇外翻,以及耳廓缺如和末节指(趾)骨坏疽。Kelsell等(2005)确认致病基因ABCA12位于2q35,并发现在12个HI中有11个患者有基因内缺失和移码缺失。ABCA12基因目前大约有56个突变被报道,分散在48个HI、10个LI、8个NCIE家系中。62.5%为致蛋白截断突变,引起的表型大多为HI.而NCIE的突变是至少含一错义突变,在基因编码蛋白第一个与ATP结合域发生的复合错义突变会引起LI[11]。ARCI致病基因较多。Fischer等(2009)[12]在对520个独立家系队列研究中认为,ARCI致病基因32%为TGM1;16%为Ichthyin,12%为ALOX12B;8%为CYP4F22;5%各为ALOXE3和ABCA12。大约22%没有发现基因突变。近来NIPAL4基因突变在ARCI也被报道。迂回线状鱼鳞病即Netherton综合征(NS)。临床特征:患者生后不久出现呈匐行性环状或多环状鳞屑性损害,皮疹呈湿疹样或脂溢性皮炎样;具有特应性素质;竹节样毛发。目前致病基因为SPINK5基因SPINK5基因位于染色体5,基因编码淋巴上皮Kazal型相关的抑制蛋白。Chavanas(2000)将NS致病基因定位在5q32。Walley等(2001)发现能编码的多态G1258A与特应性皮炎、哮喘有关。Kato等(2003)将8个多态在日本对124个AD患者和110个对照作了研究。发现包括G1258A在内的7个多态与AD是相关的;但血清IgE水平与SPINK5基因型没有相关。Zhao等(2012)[12]也发现多态G1258A,G2475T在中国东北人群中与AD相关。在表型与基因型研究中,Komatsu等(2008)[13]认为SPINK5基因突变发生的部位越早,LEKTI蛋白表达也就越少,表型也越重。出生时身体情况、生长滞后、裸露部皮肤易激惹及皮肤感染的发生频度被认为与基因型是有关的;瘙痒、嗜酸性粒细胞和IgE水平、竹节发、汗液分泌则无关。X连锁鱼鳞病X连锁隐性遗传病(XLRI),该病发生在男性,女性仅为携带者。常于出生时或生后不久即发病,表现为全身皮肤干燥、粗糙,覆着黑褐色鳞片,主要累及肢体伸侧颈部及耳前区受累为该病的特征。目前致病基因为STS基因。STS基因位于染色体Xp22.3。此基因与Y染色体有同源序列,即存在假基因。基因编码类固醇硫酸酯酶(STS)。STS在表皮主要位于颗粒层板层体,在角质层的间隙中产生胆固醇起到屏障作用。随着胆固醇硫酸盐降解累积到一定程度,桥粒降解,导致正常的脱屑。STS基因cDNA是Ballabio等从胎盘组织克隆的。STS基因突变一般可分为:完全缺失、部分缺失、点突变。所有突变都导致STS酶活性降低。Cañueto等(2010)[14]采用扩增外显子1、5、10方法研究40个XLRI,其中完全缺失占90%,远高于既往报道。目前报道的13个点突变大都位于外显子5-10,即编码STS酶的羧基端部位。STS基因所在区域里有多个其它基因,当出现全部缺失或部分缺失时相应基因也会影响,这就是所谓毗连基因缺失综合征。最常影响的基因为VCX、VCX3A。毗连基因缺失综合征不仅有XLRI症状而且还有如隐睾、骨骼异常、单侧肾发育不全、智力发育障碍等更加严重症状(Kallman综合征)[15]。参考文献1.NomuraT,SandilandsA,AkiyamaM,etal.UniquemutationsinthefilaggringeneinJapanesepatientswithichthyosisvulgarisandatopicdermatitis.JAllergyClinImmunol.2007,119(2):434-440.2.ZhangH,GuoY,WangW,etal.MutationsinthefilaggringeneinHanChinesepatientswithatopicdermatitis.Allergy.2011,66(3):420-427.3.HuZ,XiongZ,XuX,etal.Loss-of-functionmutationsinfilaggringeneassociatewithpsoriasisvulgarisinChinesepopulation.HumGenet.2012,131(7):1269-1274.4.LiuP,YangQ,WangX,etal.Identificationofageneticlocusforichthyosisvulgarisonchromosome10q22.3-q24.2.JInvestDermatol.2008,128(6):1418-1422.5.NishizawaA,ToyomakiY,NakanoA,etal.AnovelH1domainmutationinthekeratin2geneinaJapanesefamilywithichthyosisbullosaofSiemens.BrJDermatol.2007,156(5):1042-1044.6.BygumA,VirtanenM,BrandrupF,etal.GeneralizedandNaevoidEpidermolyticIchthyosisinDenmark:ClinicalandMutationalFindings.ActaDermVenereol.2012,doi:10.2340/00015555-1447.7.AkiyamaM,SawamuraD,ShimizuH.Theclinicalspectrumofnonbullouscongenitalichthyosiformerythrodermaandlamellarichthyosis.ClinExpDermatol.2003,28(3):235-240.8.FarasatS,WeiMH,HermanM,etal.Noveltransglutaminase-1mutationsandgenotype-phenotypeinvestigationsof104patientswithautosomalrecessivecongenitalichthyosisintheUSA.JMedGenet.2009,46(2):103-111.9.EcklKM,deJuanesS,KurtenbachJ,etal.Molecularanalysisof250patientswithautosomalrecessivecongenitalichthyosis:evidenceformutationhotspotsinALOXE3andallelicheterogeneityinALOX12B.JInvestDermatol.2009,129(6):1421-1428.10.NawazS,TariqM,AhmadI,etal.Non-bullouscongentitalichthyosiformerythrodermaassociatedwithhomozygosityforanovelmissensemutationinanATPbindingdomainofABCA12.EurJDermatol.2012,22(2):178-181.11.AkiyamaM.ABCA12mutationsandautosomalrecessivecongenitalichthyosis:areviewofgenotype/phenotypecorrelationsandofpathogeneticconcepts.HumMutat.2010,31(10):1090-1096.12.FischerJ.Autosomalrecessivecongenitalichthyosis.JInvestDermatol.2009,129(6):1319-1321.13.KomatsuN,SaijohK,JayakumarA,etal.CorrelationbetweenSPINK5genemutationsandclinicalmanifestationsinNethertonsyndromepatients.JInvestDermatol.2008,128(5):1148-1159.14.CanuetoJ,CiriaS,Hernandez-MartinA,etal.AnalysisoftheSTSgenein40patientswithrecessiveX-linkedichthyosis:ahighfrequencyofpartialdeletionsinaSpanishpopulation.JEurAcadDermatolVenereol.2010,24(10):1226-1229.15.ChoEH,KimSY,KimJK.ACaseof9.7MbTerminalXpDeletionIncludingOA1LocusAssociatedwithContiguousGeneSyndrome.JKoreanMedSci.2012,27(10):1273-1277.
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科425人已读 - 引用 着色性干皮病致病基因的研究进展和基因诊断策略
【摘要】着色性干皮病、Cockayne综合征和毛发硫营养障碍都是由于存在核苷酸损伤切除修复缺陷、不能有效修复紫外线照射引起的DNA损伤,从而出现症状的一组紫外线敏感疾病。其遗传模式均为常染色体隐性遗传模式,涉及的致病基因目前为13个。不同基因编码的各蛋白由于都处于DNA修复及转录通路上,所以各疾病之间症状时有重叠,基因型与表型关系也相当复杂。本文就这些基因近年来本研究小组及国内外的研究进展作一综述,重点阐述着色性干皮病诸基因的突变研究和基因型表型关系,以期为其基因诊断和产前诊断提供帮助。【关键词】着色性干皮病;Cockayne综合征;毛发硫营养障碍;基因ResearchadvancesincausativegenesofXerodermaPigmentosumandrelateddiseasesSUNZhong-hui,GUOYun-yi,ZHANGJia,ZHUANGYin,LIMing,YAOZhi-rong.DepartmentofDermatology,FengxianInstituteofDermotosisPrevention,Shanghai201408,ChinaCorrespondingauthor:YAOZhi-rong,Email:zryaosmu@sohu.com【Abstract】UV-sensitivedisordersrefertoagroupofdiseasesduetothepresenceofdamagetothenucleotideexcisionrepairdefectcannoteffectivelyrepairDNAdamagecausedbyultravioletradiation.Geneticmodelofthemisautosomalrecessive,currentlyinvolving13diseasegenes,mainlyincludingxerodermapigmentosum,Cockaynesyndromeandtrichothiodystrophy.SinceeachproteinencodedbythegeneisintheDNArepairandtranscriptionpathways,thereisoverlapbetweenthesymptomsofthegroupdiseaseandgenotypephenotyperelationshipsarequitecomplex.Inthispaper,asummaryoftheresearchprogressofthesegenesinrecentyearsisreviewed,mainlyfocusingonxerodermapigmentosumgenemutationresearchsandgenotypephenotyperelationshipinordertoprovidehelpforitsgeneticdiagnosis,prenataldiagnosis.【Keywords】xerodermapigmentosum;Cockaynesyndrome;trichothiodystrophy;gene基金项目:上海市奉贤区科学技术委员会科学技术发展基金项目(奉科20131210)作者单位:201408上海,奉贤区皮肤病防治所皮肤科(孙忠辉、郭韫懿);200092上海,上海交通大学医学院附属新华医院皮肤科(张佳、庄寅、李明、姚志荣)本文主要缩写:XP:着色性干皮病(xerodermapigmentosum),CS:Cockayne综合征(CockayneSyndrome),TTD:毛发硫营养障碍(trichothiodystrophy),COFS:脑-眼-面-骨骼综合征(cerebro-oculo-facial-skeletalsyndrome),NER:核苷酸损伤切除修复(nucleotideexcisionrepair),TFIIH:转录因子IIH(transcriptionfactorIIH)为维持基因组的完整性及避免基因损害带来的负面影响,细胞具备一些化学性及遗传性的DNA修复途径。核苷酸剪切修复是最重要的DNA修复系统之一,它对消除由于致瘤药物以及紫外线照射所引起的某些DNA损伤起着关键作用。核苷酸切除修复(nucleotideexcisionrepair,NER)主要包括:①识别损伤部位;②解旋损伤部位双链;③损伤部位5′和3′端切开;④修复合成切除空隙的DNA。NER有两个途径:全基因修复(GGR)和转录偶联修复(TCR)。识别损伤的蛋白质不同是两个修复途径的主要差别。GGR中碱基损伤识别的是由两个独立的复合物DDB1/DDB2和XPC/HR23B参与;TCR中CSB介导RPA参与损伤识别[1](图1)。如果存在NER缺陷,对紫外线照射造成所致的嘧啶二聚体就不能有效清除,损伤的DNA无法修复,这将会为患者带来一系列的机能紊乱。如发育缺陷、神经畸形、光过敏、癌症以及加速老化过程。NER缺陷疾病主要包括有:着色性干皮病、Cockayne综合征和毛发硫营养障碍等,它们是一组少见的常染色体隐性遗传病。本文就这些疾病的致病基因研究进展做一概述,因限于篇幅,重点是XP诸基因的突变研究和基因型表型关系,以期为其基因诊断和产前诊断提供帮助。一、临床表现着色性干皮病(xerodermapigmentosum,XP),1870年由Kaposi等首先报道。该病是第一个被发现与损伤DNA修复缺陷有关的疾病[1],可累及各种族人群,以日本人和中东人发病率最高。XP的最主要特征是患者皮肤对阳光敏感并可在幼年发生癌变。通常在出生后最初几年发病,一开始常会在皮肤暴露部位出现炎症、溃烂、遗留色素沉着,随后几乎90%的病人会在十多岁时发生基底细胞癌、鳞状癌、黑色素瘤等皮肤癌,并可伴有智力发育迟缓、神经系统功能紊乱等症状。XP的临床症状较复杂多变,并且伴发疾病较多[2]。到目前为止,研究发现XP共有9个互补组XPA、XPB、XPC、XPD、XPE、XPF、XPG、XPH和1个变异型(XPV)。与之相对应的基因9个。互补组概念是用来进行分类,以区分DNA损伤细胞在修复通路功能上的缺陷。因为通过细胞融合技术相互融合不同XP患者的成纤维细胞形成的杂种细胞可以恢复DNA损伤修复能力,提示了XP各型之间互补。XP患者常伴有神经系统病变,约30%伴有进行性神经系统损害,可出现小头畸形、生长障碍、神经性耳聋、手足徐动症、共济失调和痉挛性截瘫等。但与Cockayne综合征不同,此类患者通常无性发育障碍。主要组织学改变为神经元变性但不伴有炎症或异常沉积。Cockayne综合征(Cockaynesyndrome,CS)临床特征表现为患者皮肤对紫外线高度敏感,而且头发柔细、皮下组织减少、皮肤干燥、少汗,类似“早老症”;神经发育异常,包括生长迟滞、精神运动发育迟缓,身高、体重、头围均落后于同年龄同性别正常儿童;眼病变表现最具有特征性的变化是色素性视网膜萎缩,可继发白内障、视神经萎缩;CS患儿的平均死亡年龄为12岁3个月。然而与XP不同的是,CS患者却鲜有癌症报道。临床分为:CSI型、CSII型和CSⅢ型。目前,已经鉴定出两个致病基因CSA与CSB。其编码蛋白也参与转录偶联修复(TCR)机制。毛发硫营养障碍(trichothiodystrophy,TTD),主要症状是硫缺乏性脆发。头发稀少、干燥、易折,其表皮细胞中严重缺乏富含半胱氨酸的蛋白质。一些TTD患者也表现为光敏感性并常伴有鱼鳞病、生长迟缓、先天性白内障等,但与CS患者一样也无易感皮肤癌。该病是由于编码DNA修复转录因子TFIIH中的XPB、XPD以及p8/TTDA三个亚基基因突变而致。XP、CS和TTD具有重叠的遗传学和临床特征。例如XP/CS重叠综合征:患者既有XP的皮肤和眼部变化,又有CS的体细胞发育、神经系统异常,因此表现为皮肤对阳光过敏并伴有色素沉着变化、视网膜变性、而且身材矮小、性发育不成熟。由于核苷酸损伤切除修复缺陷疾病有重叠,故目前该类疾病分有着色性干皮病(XP),XP伴有神经系统病变,CS综合征,XP/CS重叠,毛发硫营养障碍(TTD),XP/TTD重叠,脑-眼-面-骨骼综合征(COFS),COFS/TTD重叠,CS/TTD重叠和紫外线敏感综合征(UVSS)等10种。涉及致病基因13个。XP致病基因为:XPA、ERCC1、ERCC3(XPB)、XPC、ERCC2(XPD)、DDB2(XPE)、ERCC4(XPF)、ERCC5(XPG)和POLH。CS致病基因为:ERCC6(CSB)、ERCC8(CSA)。TTD致病基因为:ERCC3(XPB),ERCC2(XPD),GTF2H5(TTD-A)和C7orf11(TTDN1)。二、基因型与表型相关性研究核苷酸损伤切除修复缺陷疾病在基因型与表型关系的共性上有三大特点。1.遗传异质性:由于各基因突变都会影响NER,所以临床症状较为相似。例如早期XP各互补组患者皮肤暴露部位出现炎症、溃烂、遗留色素沉着。2.具有多效性:同一基因不同突变可导致不同的临床表型。比如XPB和XPD,都会导致CS、TTD、COFS。3.临床异质性:即同一突变可引起不同表型。在同一家系可解释为延迟显性。但在不同家系,后天的环境也极为重要。由于该类疾病致病基因众多无法一一罗列,下文仅将重点介绍XP各互补组。XPA互补组XPA基因位于9q22.3,含6个外显子。XPA蛋白由273个氨基酸构成,在体内XPA蛋白与复制蛋白A(RPA)结合构成杂二聚体参与损伤DNA的识别(图1)。XPA互补组患者大约占XP25%。患者多见于日本。目前为止,人类基因突变数据库HGMD(http://www.hgmd.cf.ac.uk/)中基因致病的突变类型有:错义突变/无义突变(n=12)、剪切突变(n=9)、调节突变是啥?(n=1)、小片段缺失(n=6)、小片段插入(n=3),小片段插入缺失(n=1),共计32种。研究显示XPA组的病人除表现出对皮肤的损伤外,还常伴中枢和外周神经系统异常。在日本80%的XPA患者是(IV3-1G>C)即发生在内含子3区间与外显子4接头处的剪切突变。该突变的患者如为纯合,则会有严重的、渐进的神经变性。外显子6的无义突变Arg228X在突尼斯人群中常见,有此突变的患者症状较轻[2]。另外二个较常见突变是在外显子3的无义突变Y116X和R228X。XPA热点突变区在3-5外显子区。它编码的蛋白是DNA结合区。在此区域发生的常为纯合突变,表型较严重。而表型较轻的至少有一个杂合突变发生在外显子6。此区编码TFIIH。通过广泛比较XPA各种突变及其编码的蛋白功能区和临床症状严重度,发现C端在整个蛋白中功能较为不重要[3]。在日本剪切突变IV3-1G>C是建立者突变(foundermutation)。有100万(约占人口1%)为杂合的携带者,他们在临床上并不发病。此突变纯合患者比含该突变的复合杂合患者症状严重[3]。XPC互补组XPC基因位于3p25,含有16个外显子,编码940个氨基酸的蛋白。XPC细胞仅有GGR缺陷,而TCR正常。XPC蛋白与hHR23B蛋白形成复合物参与损伤DNA的识别(图.1)。XPC互补组患者也大约占XP的25%。患者多见于美国和欧洲。目前XPC基因致病的突变类型有:错义突变/无义突变(21)、剪切突变(7)、调节突变(2)、小片段缺失(16)、小片段插入(3),小片段插入缺失(1)、大片段缺失(2)、大片段插入(1)、复杂重排(2),共计55种。研究发现c.1643_1644delTG在北非摩洛哥是高频突变[4]。Schafer分析16个XPC患者认为表型的差别是光照引起[5]。建立者突变,非洲马约特[6]有(IVS12-1G>C),在北非埃尔及利亚、突尼斯是V548AfsX572[7],Val548AlafsX25[8]。研究发现具有剪切突变的XPC患者表型轻重不一。细胞检测XPCmRNA轻微表型的仅有正常的3%,而严重表型的未能检测到。XPB、XPD互补组XPB、XPD蛋白共同参与损伤部位双链的打开。这两组患者细胞的TCR、GGR完全缺陷。XPB蛋白、XPD蛋白是转录因子TFIIH的两个亚基,都是ATP依赖的解链酶。XPB(ERCC3)位于2q14.3,含有15个外显子,编码782个氨基酸的蛋白。该蛋白具有3ˊ-5ˊ解链酶活性。作为TFIIH的一部分,XPB蛋白参与DNA的转录、修复。它是生命所必需的一个解链酶,因而这组患者罕见。基因致病的突变类型有:错义突变/无义突变(7)、剪切突变(2)、小片段缺失(1)、小片段插入(1),小片段插入缺失(1),共计11种。其中4种与XP/CS有关,2个与XPB有关,另外T119P是TTD致病突变。目前仅有六个家系,其中剪切突变Q739insX42最常见。XP/CS患者中错义突变症状较轻。而无义突变症状较重,有严重神经系统受累[9]。XPD(ERCC2)位于19q13,含有23个外显子,编码760个氨基酸的蛋白,该蛋白具有5ˊ-3ˊ解链酶活性。XPD患者临床表现严重,皮肤癌的发生率较高,伴有神经系统异常,一般发生在20岁左右。XPD互补组患者大约占XP的15%。基因致病的突变类型有:错义突变/无义突变(41)、剪切突变(5)、小片段缺失(10)、小片段插入(1),小片段插入缺失(1)、大片段缺失(2),共有60种.XPB,XPD也是TTD的致病基因。临床上同样这两基因引起的XP光敏明显,且易患肿瘤。尽管同样基因存在缺陷,但是细胞学研究显示TTD患者细胞NER功能却正常。而且患者并没有皮肤肿瘤。神经影像检查表明,TTD和CS患者是神经髓鞘形成不良而不是脱髓鞘。TTD患者的神经系统反常是此发育缺陷引起,所以进行性神经变性在TTD患者中未见报道。从基因突变分析光敏型TTD大多位于R112环和蛋白C末端[10]。具有XPD突变患者可显示出遗传异质性。而来自于XPD突变个体的细胞,经常发现错义突变会导致蛋白残留一些功能。ERCC2基因突变的XP患者DNA损伤处有持续NER蛋白的累积,而TTD患者却无累积[11]。XPE互补组XPE患者的日光敏感程度最轻。XPE细胞存在GGR缺陷,而TCR正常。XPE细胞缺乏紫外线损伤的DNA结合蛋白(UV-DDB)活性。UV-DDB是由p127、p48两个亚单位构成的杂二聚体,参与损伤DNA的识别。这两个亚单位分别由DDBl、DDB2编码(图1)。其中DDB2基因位于11p12,有10个外显子,编码427氨基酸的p48。XPE患者的UV-DDB活性缺失是由DDB2基因突变所致,DDB1突变患者至今尚无报道。基因致病的突变类型有:错义突变/无义突变(7)、剪切突变(1)、小片段缺失(2),共有10种。研究显示发生XPE突变的患者罕见,目前大约有十几例患者报道。DDB2基因突变都造成蛋白截断和内部缺失。这些缺陷导致P48蛋白表达严重下降,以致不能与P127亚基相互作用,从而造成UV-DDB结合活性缺乏。Oh等分析了4例XPE突变的患者并总结既往8例,发现XPE成人患者都有各种大量的皮肤肿瘤,从而推测DDB2基因突变与皮肤肿瘤发生有关[12]。XPF互补组ERCC4(XPF)基因位于16p13.13,含有11个外显子。XPF蛋白由905个氨基酸组成,XPF蛋白和ERCCl蛋白单独存在是不稳定的。XPF蛋白N末端的378个氨基酸具有核酸内切酶活性,与ERCCl蛋白相互作用形成稳定的杂二聚体XPF-ERCCl,发挥5’核酸内切酶活性。杂二聚体中的ERCCl蛋白又与XPA蛋白相互作用,该蛋白引导XPF核酸内切酶准确定位。XPF互补组患者大约占XP的6%。基因致病的突变类型有:错义突变/无义突变(15)、调节突变(1)、小片段缺失(4)、小片段插入(1),大片段缺失(1),共有22种.XPF组的病人虽对紫外线敏感但病情却较轻。在XPF突变可能有轻微的疾病或成人时发病才有严重的神经系统变性。研究发现,在XPF的错义突变可以不仅导致XP,也会造成一种早衰综合征。在体外和体内,Ahmad等[13]对引起早老症的突变R153P和引起XP的突变R799W的进行了效果比较。试图确定XPF突变如何可以导致这些不同的症状。他们发现源于R153P的细胞质中XPF-ERCC1丰富,而导致XP的细胞质XPF-ERCC1却仅有小部分。这表明,至少DNA修复缺陷和突变的相关症状的一部分是由于XPF-ERCC1的错误定位引起。XPG互补组ERCC5基因位于13q33.1,含有18个外显子。编码1186氨基酸,该蛋白具有3’核酸内切酶活性。损伤修复需要XPG蛋白发挥其核酸内切酶活性,而转录只需其特异地结合到DNA上,无需内切酶活性。XPG基因的突变可以导致XP和CS综合征。XPG基因突变的位置、类型决定所致疾病的类型。XP是由于XPG基因点突变引起核酸内切酶活性缺陷所致,CS综合征则是由于XPG基因缩短突变引起转录活性缺陷所致。XPG互补组患者大约占XP6%。基因致病的突变类型有:错义突变/无义突变(16)、小片段缺失(7)共计23种.XPG造成蛋白截断的突变主要出现于XP/CS患者,而XP-G错义突变出现于无神经系统疾病的XP患者,这些错义突变保留了一些XPG蛋白活性[14]。迄今为止报道的约16例XPG突变患者中7例为CS综合征,2例为伴有神经系统疾病的XP,余者为无神经系统疾病的XP[14]。XPH互补组ERCC1该基因位于19q13.32,含有10个外显子。编码1493碱基的蛋白,功能是形成稳定的杂二聚体XPF-ERCCl,用于切割损伤的核苷酸片段5’端。致病的突变类型有:错义突变/无义突变(2)、调节突变(3)、)仅5种。尽管此基因在修复基因中最早被克隆,但目前在人类仅有一例报道,其余是在哺乳动物病例上发现。Jaspers等[15]发现的是一复合杂合突变。表现的NER损伤效应类似XPF突变,但是该病例在婴儿早期即因发育衰竭而死亡。XPV互补组POLH基因位于6p21.1,含有11个外显子。编码713个氨基酸组成的DNA聚合酶eta,该蛋白功能与错配修复有关。XPV细胞无NER缺陷,但存在跨损伤合成(TranslesionSynthesis,TLS)缺陷。XPV互补组患者大约占XP的21%。患者多见于美国和欧洲。XPV患者尽管有各种皮肤肿瘤但是没有神经系统损害。基因致病的突变类型有:错义突变/无义突变(23)、剪切突变(2)、小片段缺失(9)、小片段插入(5)、大片段缺失(4)、大片段插入(1),共有44种。XPV在日本也较多见。Tanioka[16]研究16个日本XPV患者显示皮肤肿瘤平均发病年龄为45岁,而欧美患者在70岁以上。该研究还显示在检测到的突变中,G490T(占39%)是一高频突变。目前XPV没有发现明确的基因型表型关系。通过总结文献报道的XPV患者得出可能的假设是:基因突变如发生在POLH催化结构域外,症状总是较轻[17]。Cockayne综合征ERCC6(CSB)基因位于10q11.23,21个外显子。编码1493个氨基酸的蛋白。致病的突变类型有:错义突变/无义突变(35)、剪切突变(11)、调节突变(1)、小片段缺失(13)、小片段插入(7)、大片段缺失(6)、大片段插入(1),共计74种。ERCC8(CSA)基因定位于5q12.1,12个外显子。编码396个氨基酸的蛋白。致病的突变类型有:错义突变/无义突变(11)、剪切突变(7)、小片段缺失(1)、小片段插入(2)、小片段插入缺失(1)、大片段缺失(4)、大片段插入(1)、复合突变(1)共计28种.CS综合征中大约有65%的是ERCC6基因突变。两基因引起的表型是非常相似的,但是症状严重的CSⅡ型是由ERCC6基因引起。尽管许多家系有特定突变,但在某些人群还是有建立者突变。ERCC6基因:索马里Gly184AspfsX28,日本Asp93LeufsX26、巴西Val105ThrfsX6、讲葡萄牙语的巴西人Ala207_Ser209del、北非Tyr200LysfsX12。ERCC6基因:法国Glu182AsnfsX4、英国Phe665_Gln723、留尼旺岛有5’端不翻译区碱基缺失。ERCC8基因中所有错义突变都位于编码蛋白的WD模体(motif)区,其中在第四个模体占所有CSA错义突变的一半。ERCC6基因,错义突变大都集中在蛋白的保守区670和687之间[18]。CS基因型表型关系还不明确。在CSB,一个镶嵌在CSB基因中能编码PGBD3转座子(transposon)的基因内含子5区最近受到研究。目前认为,由CSB前5个外显子和PGBD3转座子组成的融合蛋白导致CSB蛋白不能表达,从而出现CS表型。作者猜测,内含子5的下游突变会导致表现严重的CS,而上游则会较轻[18]。三、结语核苷酸损伤切除修复缺陷疾病是一组的常染色体隐性遗传疾病。由于种族在人文、地理环境上存在差异,因而在常见基因型、表型上都有所差异。尤其体现在XP,许多种族都有建立者突变。目前在中国人群对XP的基因诊断开展还较少,同样对其基因型表型关系也缺乏研究。文献中,杨勇教授在2004年最先产前诊断了一例XPC[19],其余报道分别为2例XPC[20]和1例XPV[21]。本课题组近期研究了12个汉族XP家系、13个患者,仅发现了XPA、XPC、XPG和XPV4个互补组[22]。由于存在众多XP致病基因,为高效经济的基因诊断临床可疑XP患者,结合我们的研究结果及以上概述的XP及相关疾病致病基因的研究进展,我们提出目前汉族人群的XP基因诊断策略。如果一个临床诊断的XP患者伴神经系统损害,那么首先检测XPA的致病基因,然后XPC、XPG和XPV,假如XP患者未伴有神经系统损害,检测顺序则正好相反。若XPAXPC、XPG和XPV基因突变分析都为阴性,最后检测剩余4型(XPBXPD、XPE和XPF)总之,本文概述了核苷酸损伤切除修复缺陷疾病致病基因研究进展,重点是XP诸基因的突变研究和基因型表型关系,目的是为其基因诊断、产前诊断提供帮助。参考文献1.GuoC,TangTS,FriedbergEC.SnapShot:nucleotideexcisionrepair.Cell,2010,140(5):754.2.MessaoudO,BenRekayaM,KefiR,etal.IdentificationofaprimarilyneurologicalphenotypicexpressionofxerodermapigmentosumcomplementationgroupAinaTunisianfamily.BrJDermatol,2010,162(4):883-886.3.TakahashiY,EndoY,SugiyamaY,etal.XPAgenemutationsresultinginsubtletruncationofproteininxerodermapigmentosumgroupApatientswithmildskinsymptoms.JInvestDermatol,2010,130(10):2481-2488.4.SenhajiMA,AbidiO,NadifiS,etal.c.1643_1644delTGXPCmutationismorefrequentinMoroccanpatientswithxerodermapigmentosum.ArchDermatolRes,2013,305(1):53-57.5.SchäferA,HofmannL,GratchevA,etal.Moleculargeneticanalysisof16XP-CpatientsfromGermany:environmentalfactorspredominatelycontributetophenotypevariations.ExpDermatol,2013.22(1):24-29.6.CartaultF,NavaC,MalbrunotAC,etal.AnewXPCgenesplicingmutationhasleadtothehighestworldwideprevalenceofxerodermapigmentosuminblackMahoripatients.DNARepair(Amst),2011,10(6):577-585.7.BenRekayaM,MessaoudO,TalmoudiF,etal.HighfrequencyoftheV548AfsX572XPCmutationinTunisia:implicationformoleculardiagnosis.JHumGenet,2009,54(7):426-429.8.SoufirN,GedC,BourillonA,etal.AprevalentmutationwithfoundereffectinxerodermapigmentosumgroupCfromnorthAfrica.JInvestDermatol,2010,130(6):1537-1542.9.OhKS,ImotoK,BoyleJ,KhanSG,etal.InfluenceofXPBhelicaseonrecruitmentandredistributionofnucleotideexcisionrepairproteinsatsitesofUV-inducedDNAdamage[J]..DNARepair(Amst),2007,6(9):1359-1370.10.HashimotoS,EglyJM.TrichothiodystrophyviewfromthemolecularbasisofDNArepair/transcriptionfactorTFIIH.HumMolGenet,2009,18(R2):R224-R230.11.BoyleJ,UedaT,OhKS,etal.PersistenceofrepairproteinsatunrepairedDNAdamagedistinguishesdiseaseswithERCC2(XPD)mutations:cancer-pronexerodermapigmentosumvs.non-cancer-pronetrichothiodystrophy.HumMutat,2008,29(10):1194-1208.12.OhKS,EmmertS,TamuraD,etal.MultipleskincancersinadultswithmutationsintheXP-E(DDB2)DNArepairgene.JInvestDermatol,2010,131(3):785-788.13.AhmadA,EnzlinJH,BhagwatNR,etal.MislocalizationofXPF-ERCC1nucleasecontributestoreducedDNArepairinXP-Fpatients.PLoSGenet,2010,6(3):e1000871.14.MoriwakiS,TakigawaM,IgarashiN,etal.XerodermapigmentosumcomplementationgroupGpatientwithanovelhomozygousmissensemutationandnoneurologicalabnormalities.ExpDermatol,2012,21(4):304-307.15.JaspersNG,RaamsA,SilengoMC,etal.FirstReportedPatientwithHumanERCC1DeficiencyHasCerebro-Oculo-Facio-SkeletalSyndromewithaMildDefectinNucleotideExcisionRepairandSevereDevelopmentalFailure.AmJHumGenet,2007,80(3):457-466.16.TaniokaM,MasakiT,OnoR,etal.MolecularanalysisofDNApolymeraseetageneinJapanesepatientsdiagnosedasxerodermapigmentosumvarianttype.JInvestDermatol,2007,127(7):1745-1751.17.InuiH,OhKS,NademC,etal.Xerodermapigmentosum-variantpatientsfromAmerica,Europe,andAsia.JInvestDermatol,2008,128(8):2055-2068.18.Laugel,V.Cockaynesyndrome:Theexpandingclinicalandmutationalspectrum.MechAgeingDev,2013,134(5-6):161-170.19.YangY,DingB,WangK,etal.DNA-basedprenataldiagnosisinaChinesefamilywithxerodermapigmentosumgroupA.BrJDermatol,2004,150(6):1190-1193.20.LamCW,CheungKK,LukNM,etal.DNA-baseddiagnosisofxerodermapigmentosumgroupCbyWhole-genomescanusingsingle-nucleotidepolymorphismmicroarray.JInvestDermatol,2005,124(1):87-91.21.LiuX,ZhangX,QiaoJ,FangH.IdentificationofanovelnonsensemutationinPOLHinaChinesepedigreewithxerodermapigmentosum,varianttype.IntJMedSci,2013,10(6):766-770.22.SunZ,ZhangJ,GuoY,etal.Genotype-phenotypecorrelationofxerodermapigmentosuminaChineseHanpopulation[J].BrJDermatol,2015,172(4):1096-1102.23.DiGiovannaJJ,KraemerKH.Shiningalightonxerodermapigmentosum.JInvestDermatol,2012,132(3Pt2):785-796.
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科313人已读 - 遗传性皮肤病
人类皮肤及其附属器的结构和功能的差异大多是遗传因素所决定,所以许多皮肤病遗传性因素在发病机制上起很大作用。遗传性皮肤病中有的仅主要表现为皮肤的异常,也有许多是伴有皮肤异常的系统疾病。 根据皮肤病的遗传方式,分为:一、常染色体隐性遗传,一般双亲为正常,但其兄弟姐妹可能患病是这类遗传的特点,双亲家属发病率较高,患者的智力等一些功能会有明显性的障碍,看不到连续几代遗传,生命预后差等特点。如迂回线状鱼鳞病,着色性干皮病。二、常染色体显性遗传,这类皮肤病的特点是双亲中至少有一个是患者,子女至少有一半患病,和性别不会有多大的关系,通常病情不会很严重,并且不会影响到生命和正常的工作能力。常染色体显性遗传性皮肤病占遗传性皮肤病的70%左右,常见的有寻常型鱼鳞病、家族性慢性良性天疱疮、汗孔角化症、神经纤维瘤病等。三、多基因遗传,这类皮肤病是指遗传特征是由几对基因所决定的,而不单单只是一对基因,并且由几对基因所决定的遗传方式,环境因素对患者的影响也回比较大,家族中发病率明显高于一般群体的特点。多基因遗传性皮肤病常见的有:脂溢性皮炎、寻常痤疮、红斑狼疮、银屑病、多毛症、斑秃等。四、X性联遗传,此类皮肤病的特点是男性发病率要高于女性,并且是隔代遗传,女患者所生儿子全部发病。如性联遗传性鱼鳞病。症状体征遗传性皮肤病由亲代将其本身的异常或突变后的基因传给下一代,从而使下一代出生时或以后出现该基因所特有的皮肤损害。非遗传性的先天性皮肤病为数很少,主要是胎儿在母体内发育过程中受到营养、病毒或微生物感染,或化学、物理因素的干扰而引起的皮肤及其附属器的疾病。①遗传性皮肤病的一般特点同遗传性疾病;②可以生后即有,也可在儿童或青春期出现;③其症状不一定为该遗传性皮肤病所特有,例如摩擦引起的大疱与先天性大疱性表皮松解症者相似;④同一种疾病在不同人表现的症状轻重不同,例如鱼鳞病轻者仅四肢有轻微改变,重者躯干、四肢均可累及,并且角化干燥突出,还可伴发毛囊角化、掌跖角化等;⑤同一种临床表现可有不同的遗传方式,例如鱼鳞病可有不同遗传模式引起。治疗多数无有效疗法,不能根除。个别疾病可通过药物或饮食治疗得到改善,例如遗传性血管性水肿是由常染色体显性遗传的C1酯酶抑制剂缺乏所致,可给血管舒缓素和雄激素样药物达那唑治疗,苯丙酮酸尿症则限制饮食中苯丙氨酸的摄入即可不发病;肠病性肢端皮炎可通过给锌制剂加以控制;有些先天性酶缺陷或丙种球蛋白缺乏,可通过补充缺少的有关物质进行治疗。但这些治疗并不能去除病因。有些先天性畸形或异常影响功能或美容时,可根据具体情况采取手术治疗。疾病诊断目前基因诊断是遗传性皮肤病的金标准。基因诊断需注意三点:病史:遗传性皮肤病多种多样,临床表现各不相同,但除每种具体疾病所特有的体征外,还有以下共性特点,可供诊断参考:①绝大多数在出生时或在婴儿、儿童期表现症状;②父母及其上代或兄弟姐妹有同样疾病;③父母为近亲结婚;④皮肤体征明显,但缺少主观症状。有的表现为多系统疾病;⑤长期存在,不易治疗;样本:可以是任何有核细胞,包括:外周血白细胞、口腔黏膜细胞、活检标本、石腊包埋的组织块、沉淀细胞(唾液、痰液、尿液)、羊水细胞、绒毛细胞、进入母体循环的胎儿细胞诊断基本技术:核酸杂交、聚合酶链反应、DNA测序、基因芯片技术预防保健本病不能通过药物或其他手段根治,故预防更为重要。对已有本病者,应避免接触有害因素预防或减少其症状发生。例如着色性干皮症患者应避免日晒,大疱性表皮松解症应避免摩擦与压迫,以减少症状发生。另外,通过遗传咨询和产前诊断可减少本病的发生。
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科33人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科33人已读 - 黄褐斑
黄褐斑是一类常见的原因未明的、淡褐色至深褐色色素沉着病。一般发生于面部、颈部等曝光部位,以女性多见,特别是妊娠期第2〜5个月,有时也可见于绝经期妇女或男性。本病病因尚未完全明确,一般认为与遗传、女性激素有关,也与一些慢性病有关如女性生殖器疾患,内脏肿瘤、慢性酒精中毒、肝脏病、甲亢、结核以及某些药物(如避孕药、抗癫痫药等)、化妆品、日晒有关。皮损多对称分布于面部的突出部位,以颧部、前额和两颊最明显,鼻及颧部皮损融合成蝴蝶状,颜色为淡褐色至淡黑色,皮疹为大小不等、形状不规则的斑疹或斑片,表面光滑,有融合倾向,边缘较清楚表面无鳞屑;皮损面积和颜色深浅随日晒、体内激素水平变化等因素而变化。治疗方面,应在医生指导下,可选用白降汞软膏,双氧水、氢醌霜等外用。有人用维生素E霜白天外搽,维A酸霜晚上外搽,有较好效果,部分患者选用铒激光有较好疗效。中药方面,可以根据症候选用六味地黄丸、逍遥丸等治疗。预防保健1. 避免日晒,在太阳底下应打伞、涂防晒霜。2. 注意有些药物会加重皮损,特别是有些药物和食物有光3. 本病会反复发作。4. 注意有些药物有副作用,不能为追求药效盲目用药。5. 中医中药需根据症候应用,在医生指导下用药。
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科194人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科194人已读 - 白癜风
白癜风是后天色素脱失的皮肤病,可发生于任何年龄,病程长期慢性。病因不明确,有较明显遗传倾向,有的专家认为与糖尿病、恶性贫血、甲状腺病等有关系,也有学者认为与精神创伤有关。本病的临床表现为病损为大小不等局限性脱色斑,数目不定,边界清楚,周围色素可正常或加深,发病部位处毛发可变白(头发可白),无瘙痒疼痛等自觉症状,全身各部位均可发生,但以面、颈、手背多见。治疗方面,本病治疗方法很多,目前暂无特效疗法,临床一般选择联合治疗。局部治疗可用补骨脂酊、氮芥酒精、卡泊三醇软膏、他卡西醇软膏、激素外用或局部注射、光疗、表皮移植等方法;他克莫司软膏及吡美莫司乳膏可用于局限性儿童白癜风的治疗。快速进展期的儿童白癜风皮损可采用口服小剂量激素治疗。中医治疗方面,可使用白驳丸。预防保健1. 本病不具有传染性。发作期间不吃酒类等刺激食物。2. 炒菜可用铜锅,少吃酸性食物。3. 脸部用药要注意药物副作用。4. 应到正规医疗机构治疗。
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科190人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科190人已读 - 脂溢性角化
脂溢性角化病又称“老年疣”,是一种良性表皮内肿瘤。本病多见于40岁以后人群,男性更多见,随年龄增大而皮损增多。皮损初发最常见于面部、头皮、躯干、上肢,亦可累及体表任何部位,但不累及掌、跖。早期为小而扁平丘疹或斑片,淡褐或深褐色,表面光滑;逐渐增大表面干燥粗糙,底部呈圆形,椭圆形或不规则形,可形成一层油腻性厚痂,境界清楚。本病可单发,但通常多发,数目不等,多为20~40个,个别可多达百个以上。不典型者很容易与黑素瘤、色素型基底细胞上皮瘤、日光性角化病及色素痣等相混淆,若诊断不明确,必要时可活检。病程通常缓慢,一般无自觉症状,偶有痒感。本病一般不需要治疗。如觉得影响美观,可以用手术、激光、冷冻等治疗。预防保健1. 平时应避免摩擦外伤。2. 本病无自愈倾向。但不影响健康,不传染。4. 本病一般不会恶变,患者不需紧张。5. 手术切除后可能还会再长。
 孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科522人已读
孙忠辉 主任医师 上海市奉贤区皮肤病防治所 皮肤科522人已读