先天性糖基化障碍(CDG)
- 精选 先天性糖基化障碍(CDG)治疗新进展
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生自从1981年发现第一例先天性糖基化障碍患者到现在已经发现的先天性糖基化障碍类型及相关基因均超过160种(图1)。本文详细描述近年来各种先天性糖基化障碍针对性治疗措施方面的新进展,包括针对器官系统(神经系统、胃肠道、肝脏、血液系统、内分泌及免疫系统)异常的治疗措施、食物补充剂或饮食(半乳糖、甘露糖、岩藻糖、唾液酸、尿苷、金属制剂及核糖醇)治疗措施、器官移植及干细胞移植、分子伴侣药物治疗、活化的单糖药物治疗及基因治疗。本文也会描述NGLY1缺陷病(NGLY1先天性去糖基化障碍)及糖基磷脂酰肌醇锚(GPIanchor,GPI锚)相关先天性糖基化障碍的治疗措施。一、针对器官系统异常的治疗1、神经系统异常的治疗近期报道显示,PMM2先天性糖基化障碍患者乙酰唑胺治疗可以改善神经系统症状。肌肉关节强直或挛缩的患者可以考虑局部注射肉毒素。脱水、感染或发热是可能会加重癫痫、肌阵挛或其他神经系统症状,也有可能诱发脑中风样发作,需要积极治疗脱水、感染及发热。如果出现脑中风样发作,需要排查诊治脑血栓。脑中风样发作时精氨酸治疗没有看到效果,目前有效的措施是积极补液。脑中风样发作出现的单侧肢体无力多数情况下2周之内自行缓解。丙戊酸治疗通常可以改善肌阵挛症状,但肝功能异常的患者需要检测肝功能指标变化。也可以选用左乙拉西坦、布瓦西坦及氯硝西泮等药物。难治性癫痫是可以考虑生酮饮食治疗的,但糖基化障碍患者存在低血糖的风险,生酮饮食治疗需要谨慎,密切监测血糖。SLC39A8先天性糖基化障碍(SLC39A8-CDG)患者口服半乳糖及锰制剂治疗后癫痫状况有所改善。口服岩藻糖治疗后SLC35C1先天性糖基化障碍(SLC35C1-CDG)患者神经系统发育有所改善。SLC35A2先天性糖基化障碍(SLC35A2-CDG)患者口服半乳糖治疗后癫痫发作好转。补充尿苷治疗可以改善CAD先天性糖基化障碍(CAD-CDG)出现的严重癫痫,乙酰化的尿苷补充剂吸收更好,已经有FDA批准的三醋酸尿苷(Xuriden,uridinetriacetate)可以试用。DPAGT1先天性糖基化障碍(DPAGT1-CDG,旧称先天性糖基化障碍Ij型)是负责编码长萜醇磷酸N-乙酰葡萄糖胺-1-磷酸转移酶(dolicholphosphateN-acetylglucosamine-1-phosphotransferas)DPAGT1基因突变导致的常染色体隐性遗传病。严重患者出现DPAGT1先天性糖基化障碍(DPAGT1-CDG),表现为肌张力低下、白内障、骨骼异常、难治性癫痫、智力障碍、全面发育落后及早期死亡。轻度患者可以出现DPAGT1先天性肌无力综合征(DPAGT1-CMS),表现为肌无力、容易疲劳及四肢肌力降低。Özsoy等报道1例8个月龄的DPAGT1 基因纯合突变(c.339T>G/p.Phe113Leu)男婴及1例13岁DPAGT1复合杂合突变(c.466C>T/p.Arg156Cys及c.161+5G>A)的女孩。8月男婴在1个月龄是发现白内障,特殊面容及癫痫。13岁女孩的表现出现的晚,而且主要以肌肉无力、行为异常及特殊面容为主,但没有癫痫。吡啶斯的明治疗明显改善患者肌无力症状。2、胃肠道功能异常的治疗胃肠道粘膜的蛋白高度依赖糖基化,消化道淋巴回流也一定程度上依赖糖基化,故先天性糖基化障碍患者肠胃功能异常或营养吸收障碍比较常见。口面部肌肉运动障碍、肌张力低下、胃食管返流、胃肠道动力障碍、呕吐等神经系统异常均可以导致营养不良或营养摄入障碍。MPI先天性糖基化障碍(MPI-CDG)、ALG6先天性糖基化障碍(ALG6-CDG)及PMM2先天性糖基化障碍(PMM2-CDG)均可以出现丢失蛋白型肠病。丢失蛋白型肠病患者可以考虑输注白蛋白、奥曲肽(octreotide生长抑素)、富含MCT(中链甘油三酯)的饮食。胃肠道症状的治疗措施方面建议肠胃耐受的前提下尽量增加热卡摄入,部分患者需要特殊深度水解蛋白奶粉或氨基酸奶粉喂养。严重患者可以考虑鼻饲喂养或胃造瘘喂养。MAN1B1先天性糖基化障碍(MAN1B1-CDG)患者因为有肥胖症,需要减少热卡摄入。甘露糖治疗MPI先天性糖基化障碍(MPI-CDG)患者不仅可以明显改善腹泻和丢失蛋白型肠病,也可以纠正低白蛋白血症。也有一例肝素治疗控制丢失蛋白型肠病的报道。3、肝病的治疗约22%的先天性糖基化障碍类型均可以出现不同程度的肝脏异常,包括转氨酶升高、胆汁淤积症、胆管发育异常、肝囊肿、脂肪肝、肝纤维化、肝硬化及肝衰竭。胆汁淤积症或胆管异常的患者可以利胆治疗的同时监测脂溶性维生素水平,必要时补充维生素A、D、E及K1等脂溶性维生素。先天性糖基化障碍患者胆固醇水平下降比较常见,但CCDC115-CDG、ATP6VAP1-CDG,及TMEM199-CDG胆固醇水平可能会升高,胆固醇严重升高者可以考虑降血脂的药物治疗。甘露糖治疗可以可以改善MPI先天性糖基化障碍(MPI-CDG)患者肝功能指标,但不一定能逆转肝纤维化或肝硬化趋势。部分MPI-CDG及CCDC115先天性糖基化障碍(CCDC115-CDG)患者可能需要肝移植。半乳糖治疗PGM1先天性糖基化障碍(PGM1-CDG)可以使转氨酶水平下降,也有一例胆汁淤积性肝衰竭患者通过半乳糖治疗后逆转的报道。ALG8先天性糖基化障碍(ALG8-CDG)、COG6先天性糖基化障碍(COG6-CDG)、COG7先天性糖基化障碍(COG7-CDG)、CCDC115先天性糖基化障碍(CCDC115-CDG)及ATP6AP1先天性糖基化障碍(ATP6AP1-CDG)等疾病可以表现为胆汁淤积症,可能会出现脂溶性维生素缺乏及营养吸收障碍。需要检测脂溶性维生素水平,必要时补充维生素A、D、E及K1等脂溶性维生素。营养不良者可富含MCT奶粉喂养,也可以口服补充MCT油或椰子油。4、血液系统异常的治疗先天性糖基化障碍可引起血小板膜上的糖蛋白异常及凝血因子活性降低,增加血栓形成及出血风险,尤其是PMM2先天性糖基化障碍(PMM2-CDG),MPI先天性糖基化障碍(MPI-CDG)及ALG1先天性糖基化障碍(ALG1-CDG)。血液系统异常包括动脉或静脉血栓形成、粘膜或脏器出血及脑中风样发作。也有缺血性脑中风、颅内出血及弥散性血管内凝血的病例报道。发热、脱水、长期卧床或不运动、手术或外伤等均可导致的血液循环障碍或组织损害,从而增加出血或血栓形成的风险。血栓形成时可用肝素溶栓治疗,也有用利伐沙班(Rivaroxaban)的报道。预防血栓时可以用华法林。出血时可以用新鲜冰冻血浆。口服甘露糖可以纠正MPI先天性糖基化障碍(MPI-CDG)的凝血功能异常,也可以预防出血或血栓形成,也有报道肝移植后凝血功能自行恢复正常的案例。口服半乳糖也可以改善PGM1先天性糖基化障碍(PGM1-CDG)及TMEM165先天性糖基化障碍(TMEM165-CDG)的异常凝血功能指标。5、内分泌紊乱的治疗先天性糖基化障碍患者内分泌紊乱比较常见,发现的激素异常包括生长激素、甲状腺激素、胰岛素及性激素。部分营养不良或矮小的患者虽然生长激素水平正常,但是胰岛素样生长因子1(IGF-1)及胰岛素样生长因子结合蛋白3水平(IGFBP-3)下降。已报道1例PMM2-CDG患者IGF-1治疗效果良好,明显改善生长发育指标。甲状腺功能异常常见,包括THS升高、总T4和甲状腺素结合球蛋白水平下降。如果FT4水平下降,可以考虑优甲乐治疗。低血糖的患者可能会存在高胰岛素血症或肾上腺功能不足(ACTH下降),尤其是存在发热、呕吐或腹泻时容易出现严重低血糖、昏迷或抽搐。先天性糖基化障碍患者青春发育延迟比较常见。女性患者月经失调、闭经、高促性腺激素性腺功能减退、卵泡刺激素(FSH)及黄体生成素(LH)水平升高、雌激素水平下降等情况常见。也有男性患者睾丸偏小、隐睾、FSH升高等报道。甘露糖治疗后MPI先天性糖基化障碍(MPI-CDG)患者生长发育指标会明显好转,低血糖可以被纠正。半乳糖治疗可以改善PGM1先天性糖基化障碍(PGM1-CDG)患者高促性腺激素性腺功能减退表现及低血糖。同样,半乳糖治疗可以改善TMEM165先天性糖基化障碍(TMEM165-CDG)患者胰岛素样生长因子1(IGF-1)水平及胰岛素样生长因子结合蛋白3水平(IGFBP-3)。低血糖的患者是建议检查胰岛素、皮质醇、生长激素、乳酸、血氨、血酮体、游离脂肪酸及尿酮体。也建议频繁按需喂养、必要时口服补充复杂的碳水化合物或夜间持续鼻饲喂养。高胰岛素性低血糖时可以二氮嗪(diazoxide)治疗,也有切除大部分胰腺治疗高胰岛素血症的案例。性激素异常或青春发育异常的患者可以考虑小剂量雌激素替代治疗(女性)或小剂量雄激素(男性)替代治疗。6、感染及免疫功能异常治疗以下先天性糖基化障碍有明显的免疫功能异常:ALG12先天性糖基化障碍(ALG12-CDG)、MAGT1先天性糖基化障碍(MAGT1-CDG)、MOGS先天性糖基化障碍(MOGS-CDG)、PGM3先天性糖基化障碍(PGM3-CDG)、ATP6AP1先天性糖基化障碍(ATP6AP1-CDG)、ATP6AP2先天性糖基化障碍(ATP6AP2-CDG)及SLC35C1先天性糖基化障碍(SLC35C1-CDG)。典型的表现是免疫功能低下或免疫缺陷、反复或严重感染(细菌、病毒或真菌感染)、以及预防接种后产生的保护性抗体水平偏低或不产生保护性抗体。鼻窦炎、肺部感染及皮肤感染较多见。免疫功能指标异常常见的表现包括白细胞计数异常(中性粒细胞增多、中性粒细胞减少、淋巴细胞减少)及免疫球蛋白水平下降(主要是IgA及IgG)。如果CDG患者既往有反复感染或重症感染,应该详细检查免疫功能指标,出现感染时也要及时复查免疫指标。如果没有明显禁忌症,应该按时接种疫苗,接种疫苗后监测保护性抗体水平。感染时积极治疗预防败血症的发生。有报道用静脉丙种球蛋白治疗或预防感染的案例,也有预防性用抗生素的案例。SLC35C1先天性糖基化障碍(SLC35C1-CDG)是可以有用口服的特效药物(岩藻糖)治疗,治疗后反复感染及中性粒细胞计数均可以明显好转,少数患者中性粒细胞计数可以恢复正常。MAGT1先天性糖基化障碍(MAGT1-CDG)患者可以用镁制剂治疗,可以改善EB病毒血症,也可以减少EB病毒相关的血液系统恶性肿瘤的发生率。PGM3先天性糖基化障碍(PGM3-CDG)表现为IgE升高的过敏体质及自身免疫性疾病。有报道一例PGM3-CDG患者出现的严重联合免疫缺陷病通过造血干细胞移植后得以恢复。但也有报道一例SLC35A1先天性糖基化障碍(SLC35A1-CDG)患者骨髓移植后因为肺出血及移植物抗宿主反应夭折。二、食物补充剂或饮食治疗措施1、半乳糖(Glactose)治疗最初的PGM1先天性糖基化障碍(PGM1-CDG)患者接受口服的半乳糖治疗后糖基化异常得以纠正,部分临床指标也出现好转。后续更多患者接受半乳糖治疗后临床及代谢指标均有明显改善,半乳糖最大的安全剂量可以达到1.5g每公斤每日。10例SLC35A2 先天性糖基化障碍(SLC35A2-CDG)患者接受半乳糖治疗后9例患者临床症状明显改善。有报道SLC39A8先天性糖基化障碍(SLC39A8-CDG)患者接受大剂量半乳糖治疗后糖基化异常有所改善,同时大剂量补充锰制剂后癫痫症状有所好转。有报道TMEM165先天性糖基化障碍(TMEM165-CDG)患者接受半乳糖治疗后凝血功能指标有所改善,体外研究补充半乳糖后糖基化异常有所好转。2021年美国及比利时科学家联合发表多中心研究D-半乳糖治疗PMM2先天性糖基化障碍(PMM2-CDG)的疗效及安全性数据。9例患者参与研究,疗程18周,但部分轻型患者临床症状(糖基化障碍严重程度评分)及糖基化异常有改善趋势。需要更多患者随机对照研究来证明D-半乳糖能否作为PMM2先天性糖基化障碍的辅助治疗措施。已有体外研究表明甘露糖治疗ALG13先天性糖基化障碍(ALG13-CDG)的细胞或动物模型能够改善糖基化异常的报道。2、甘露糖(Mannose)治疗短期及长期补充甘露糖可以明显改善MPI先天性糖基化障碍(MPI-CDG)患者内分泌紊乱、低血糖、凝血功能障碍及肠病等临床症状,安全性良好。推荐的剂量为每次150-170(mg/kg),每日4−5 次。虽然部分患者肝纤维化指标改善,也有没能避免肝移植的患者。已有体外研究表明甘露糖治疗ALG1先天性糖基化障碍(ALG1-CDG)的细胞或动物模型能够改善糖基化异常的报道。体外试验表明PMM2基因缺陷的细胞添加D-甘露糖可纠正糖基化异常,但既往关于PMM2先天性糖基化障碍(PMM2-CDG)患者短期补充D-甘露糖研究发现疗效不佳。2021年德国科学家发布长期补充D-甘露糖在PMM2先天性糖基化障碍患者中的疗效。20例患者接受较大剂量D-甘露糖,平均治疗时间为52周(1年),治疗期间监测蛋白糖基化、血甘露糖水平及临床表现变化。经过1年多的治疗,蛋白糖基化有明显改善。治疗期间没有发现D-甘露糖的严重副作用。作者建议进一步通过随机对照,双盲的对照研究明确大剂量、长期口服D-甘露糖治疗对PMM2先天性糖基化障碍效果,也可以考虑把D-甘露糖作为辅助的治疗药物使用。3、岩藻糖(Fucose)治疗SLC35C1先天性糖基化障碍(SLC35C1-CDG)的患者岩藻糖治疗后不仅使血清岩藻糖水平迅速上升,也可以使升高的白细胞降低到正常水平。岩藻糖治疗后的患者感染次数明显减少,也改善了部分患者精神运动发育指标。有研究表明,岩藻糖治疗一对患有严重FUT8先天性糖基化障碍(FUT8-CDG)的双胞胎患者5个月后异常临床表现均有所缓解,糖基化监测显示岩藻糖化水平有所好转。定期检查肝肾功能指标均没有看到明显的副作用。作者认为FUT8-CDG患者岩藻糖治疗是安全有效的,需要更多研究来验证其疗效。FCSK先天性糖基化障碍(FCSK-CDG),旧称FUK先天性糖基化障碍(FUK-CDG)也是岩藻糖化相关的CDG,补充岩藻糖可能会改善基因功能或临床症状。GFUS先天性糖基化障碍(GFUS-CDG)是GDP岩藻糖合成障碍,可以引起发育落后、智力障碍、小头畸形、脑部畸形、喂养困难、营养不良及粗糙面容。有一例GFUS-CDG患者接受口服岩藻糖治疗(最大剂量700mg/kgd,分三次口服)后神经系统发育有所改善,营养状况有所好转,没有发现明显的副作用。4、唾液酸(Sialicacid)治疗NANS先天性糖基化障碍(NANS-CDG)唾液酸合成障碍导致的疾病,表现为神经系统异常、智力低下、骨骼发育不良及消化系统异常。曾有研究斑马鱼疾病模型中补充唾液酸后骨骼异常得以恢复。一项人体研究纳入5例NANS-CDG患者 (年龄0–28岁) 后口服补充唾液酸15个月,用药耐受性良好,产前诊断后即开始治疗的患者(胎儿期母亲补充唾液酸,出生后改用患者口服)运动发育及神经系统发育状况都好于同样的基因突变但是出生后治疗的患者。需要研究更多患者进一步证实胎儿期治疗效果是否优于产后治疗。5、尿苷(Uridine)治疗尿苷治疗CAD先天性糖基化障碍(CAD-CDG)患者可以使神经系统异常表现及血液系统异常指标迅速好转。部分CAD-CDG患者补充尿苷的同时补充半乳糖后进一步改善了糖基化异常及临床症状。部分SLC35A2 先天性糖基化障碍(SLC35A2-CDG)患者补充半乳糖的同时补充尿苷后进一步改善糖基化异常及临床症状。6、生物金属制剂治疗生物金属在细胞间粘合、能量补充、生长发育、蛋白质折叠、膜极性的维持、蛋白质/碳水化合物/脂肪代谢及糖基化等众多生物化学机制当中起到重要的作用。补充二价锰离子治疗对TMEM165先天性糖基化障碍(TMEM165-CDG)及 SLC39A8先天性糖基化障碍(SLC39A8-CDG)患者有效。2例SLC39A8-CDG患者接受每日每公斤体重15-20mg的MnSO4(硫酸锰)治疗后不仅发现糖基化异常有所改善,临床表现的好转幅度也是比较可观的。体外细胞研究表明,补充锰制剂治疗TMEM165-CDG的细胞模型以后发现糖基化异常得以纠正。锰制剂治疗也不是没有风险的,需要检测血液中锰离子浓度避免锰中毒。锰中毒的表现包括帕金森病样表现及精神症状。MAGT1 先天性糖基化障碍(MAGT1-CDG)可以引起被称之为XMEN综合征的免疫缺陷病,慢性EB病毒感染及镁离子的稳态失衡。初步的体外研究及MAGT1-CDG患者体内研究表明,补充镁制剂可以通过改善NKG2D 表达增强人体对EB病毒的抵抗力。但镁制剂治疗不是对所有接受治疗的患者都有效。7、核糖醇(Ribitol)治疗FKTN先天性糖基化障碍(FKTN-CDG)是一种福山型(Fukuyama-type)先天性肌营养不良症,表现为进行性加重的先天性肌营养不良、脑发育畸形、骨骼肌萎缩、严重智力障碍、癫痫及运动发育落后。后续可能会出现的表现可以包括肌病样面容、腓肠肌及前臂肌肉肥大及进行性加重的心脏病变。已有用口服补充的核糖醇(Ribitol)实验性治疗FKTN-CDG的报道。三、其他治疗1、器官移植及干细胞移植肝移植:有肝硬化、门静脉高压、食管胃底静脉破裂出血、肝衰竭等终末期肝病的CDG患者可以考虑肝移植。已经有MPI先天性糖基化障碍(MPI-CDG)患者成功肝移植的报道。此外CCDC115先天性糖基化障碍(CCDC115-CDG)及ATP6AP1先天性糖基化障碍(ATP6AP1-CDG)等糖基化障碍都有成功肝移植的案例。心脏移植:严重心脏受累的患者可以考虑心脏移植。数例DOLK先天性糖基化障碍(DOLK-CDG)患者已成功接受心脏移植。合并有免疫缺陷病的CDG可以考虑造血干细胞移植。已有三例PGM3先天性糖基化障碍(PGM3-CDG)患者造血干细胞移植后免疫缺陷得以缓解。2、分子伴侣药物治疗乙酰唑胺(Acetazolamide)作为分子伴侣治疗PMM2先天性糖基化障碍:近期研究发现糖基化异常可能参与小脑共济失调及脑中风样发作发病过程,乙酰唑胺(Acetazolamide)不仅可能改善其他基因突变引起的小脑共济失调症状,也可能减少脑中风样发作次数。目前已有乙酰唑胺治疗PMM2先天性糖基化障碍的临床研究注册,进一步验证乙酰唑胺能否改善PMM2先天性糖基化障碍患者共济失调症状,同时确定长期口服乙酰唑胺的安全性。3、活化的单糖药物治疗GLM101 是Glycomine公司研发的第一批孤儿药,可为人体细胞提供活化的甘露糖(甘露糖1磷酸)治疗PMM2先天性糖基化障碍,GLM101 已被美国及欧洲监管部门批准为孤儿药。目前正在进行健康志愿者用GLM101药物的安全性、耐受性及剂量探索的一期临床研究。如能够顺利完成一期临床研究,2022年下半年可能会招录PMM2先天性糖基化障碍患者进行二期临床研究。4、基因治疗利用腺病毒相关病毒(AAV)基因治疗GNE先天性糖基化障碍(GNE-CDG)取得了正常蛋白表达纠正基因突变,但仅限于小鼠模型及人类原始肌肉细胞中,需要通过体内治疗试验验证其疗效及安全性。体外反义治疗:已经分别应用于PMM2-CDG(体外成纤维细胞治疗,转录剪切的异常得以纠正)及TMEM165-CDG(体外成纤维细胞治疗,正常蛋白标的得以恢复)。其他转基因编辑治疗措施包括锌指核酶(zinc-fingernucleases)、转录激活因子样效应物核酸酶(TALENs)以及CRISPR/Cas9技术。有在其他疾病小鼠模型中研究案例,可能在不久的将来应用到先天性糖基化障碍患者或动物模型中。四、NGLY1缺陷病(NGLY1-CDDG,NGLY1先天性去糖基化障碍)的治疗有项研究发现NGLY1缺陷病果蝇动物模型出现发育落后及死亡率增加,补充N-乙酰葡萄糖胺后果蝇寿命明显延长,死亡率明显降低。无泪及泪少的NGLY1缺陷患者补充N-乙酰葡糖胺后眼泪增多。另一项药物筛查的研究发现20-羟基蜕皮激素(20E)可以部分纠正NGLY1缺陷病果蝇的全面发育落后。但是20E是昆虫特异性的发育激素,可能不能用于治疗NGLY1缺陷病的人类。基础研究发现NGLY1缺陷病可能会导致自身炎症或自身免疫状态,高通量药物筛查也发现抗炎药物、儿茶酚胺及儿茶酚胺受体激动剂等药物可能成为潜在的治疗措施。负责细胞内外转运钠离子、钾离子及氯离子的NKCC1基因可能会可能会影响NGLY1缺陷病出现的泪少或少汗等症状,NKCC1基因将来可能会成为治疗治疗NGLY1缺陷病的新靶点。电脑模拟研究NGLY1缺陷病的治疗药物(2015-2016):日本科学家发现NGLY1缺陷病小鼠及涡虫动物模型中去除ENGase基因后可以改善小鼠生存率,提示抑制ENGase酶活性可能改善NGLY1缺陷病患者病情。科学家们们通过电脑3D分子模拟研究筛查能够抑制ENGase酶活性的小分子药物,被筛查的14种通过FDA认证的药物中Prevacid(兰索拉唑)可能理论上抑制ENGase酶,Prevacid(兰索拉唑)分子结构已成为将来药物发现的参考结构。五、糖基磷脂酰肌醇锚(GPIanchor,GPI锚)相关先天性糖基化障碍治疗糖基磷脂酰肌醇(GPI)是在内质网合成的糖脂,跟蛋白结合后在高尔基体进一步加工,GPI最终起到将不同的蛋白粘附到细胞膜膜的锚。人体有150种蛋白需要与GPI锚结合,所以GPI在胚胎形成、免疫应答及神经系统发育方面至关重要。GPI锚相关疾病通常出现碱性磷酸酶升高(ALP)或降低,因为ALP通常锚定在GPI。糖基磷脂酰肌醇锚(GPIanchor)相关先天性糖基化障碍包括以下几种CDG:PIGA先天性糖基化障碍(PIGA-CDG)、PIGG先天性糖基化障碍(PIGG-CDG)、PIGL先天性糖基化障碍(PIGL-CDG)、PIGM先天性糖基化障碍(PIGM-CDG)、PIGN先天性糖基化障碍(PIGN-CDG)、PIGO先天性糖基化障碍(PIGO-CDG)、PIGQ先天性糖基化障碍(PIGQ-CDG)、PIGT先天性糖基化障碍(PIGT-CDG)、PIGV先天性糖基化障碍(PIGV-CDG)、PIGW先天性糖基化障碍(PIGW-CDG)、PGAP1先天性糖基化障碍(PGAP1-CDG)、PGAP2先天性糖基化障碍(PGAP2-CDG)及PGAP3先天性糖基化障碍(PGAP3-CDG)。补充维生素B6或活化的维生素B6(磷酸吡哆醛,pyridoxalphosphate,PLP)治疗部分GPI锚相关先天性糖基化障碍可以使惊厥症状改善。PIGO先天性糖基化障碍(PIGO-CDG)患者接受实验性的维生素B6治疗,剂量为400mg(20mg/kg),治疗后患者惊厥得以缓解。PIGM先天性糖基化障碍(PIGM-CDG)可以表现为难治性癫痫、发育落后、小头畸形及婴儿期起病的脑血栓形成。接受丁酸钠(sodiumbutyrate)治疗数周之后不仅癫痫症状显著好转,发育落后及整体状况均有明显改善,而且也没有看到明显副作用。推荐的剂量为60–90mg/kg/d,分三次口服。然而,目前尚不知丁酸钠治疗能不能改善血栓形成。此外,生酮饮食可能会改善GPI锚相关先天性糖基化障碍的癫痫症状。参考资料:FranciscoR,BrasilS,PoejoJ,JaekenJ,PascoalC,VideiraPA,DosReisFerreiraV.Congenitaldisordersofglycosylation(CDG):stateoftheartin2022.OrphanetJRareDis.2023Oct19;18(1):329.BoyerSW,JohnsenC,MoravaE.Nutritioninterventionsincongenitaldisordersofglycosylation.TrendsMolMed.2022Jun;28(6):463-481SosickaP,NgBG,FreezeHH.ChemicalTherapiesforCongenitalDisordersofGlycosylation.ACSChemBiol.2022Nov18;17(11):2962-2971. ParkJH,MarquardtT.TreatmentOptionsinCongenitalDisordersofGlycosylation.FrontGenet.2021Sep10;12:735348.VerheijenJ,TahataS,KoziczT,WittersP,MoravaE.TherapeuticapproachesinCongenitalDisordersofGlycosylation(CDG)involvingN-linkedglycosylation:anupdate.GenetMed.2020Feb;22(2):268-279GilfixBM.CongenitalDisordersofGlycosylation:APipelinetoTreatment?HumMutat.2017Feb;38(2):127. WittersP,CassimanD,MoravaE.NutritionalTherapiesinCongenitalDisordersofGlycosylation(CDG).Nutrients.2017Nov7;9(11):1222. ThielC,KörnerC.TherapiesandtherapeuticapproachesinCongenitalDisordersofGlycosylation.GlycoconjJ.2013Jan;30(1):77-84.denHollanderB,BrandsMM,deBoerL,HaaxmaCA,LengyelA,vanEssenP,PetersG,KwastHJT,KleinWM,CoeneKLM,LefeberDJ,vanKarnebeekCDM.OralsialicacidsupplementationinNANS-CDG:Resultsofasinglecenter,open-label,observationalpilotstudy.JInheritMetabDis.2023Sep;46(5):956-971. ÖzsoyÖ,CinletiT,GünayÇ,SarıkayaUzanG,YeşilmenMC,LochmüllerH,HorvathR,YişU,OktayY,HizKurulS.DPAGT1-CDG:ReportofTwoNewPediatricPatientsandBriefReviewoftheLiterature.MolSyndromol.2023Aug;14(4):322-330.
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科441人已读
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科441人已读 - 直播回放 先天性糖基化障碍与免疫功能紊乱
直播时间:2021年04月25日11:15主讲人:库尔班江·阿布都西库尔副主任医师复旦大学附属儿科医院肝病科
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科823人已读
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科823人已读 - 精选 先天性糖基化障碍(病因、分型、临床表现、诊断)
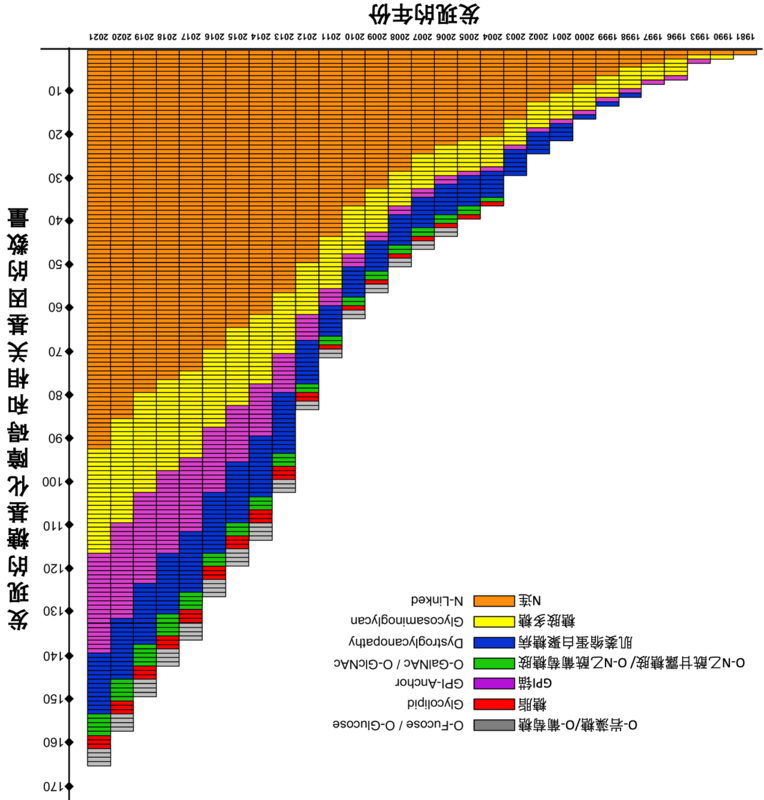
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生,王建设教授一、糖基化过程糖基化是通过酶的催化,糖和蛋白(或脂肪)结合形成糖蛋白(或糖脂)的生物化学过程。糖与蛋白结合(或脂肪)后使蛋白质(或脂肪)与相应器官组织链接,保证其功能正常运行。糖基化过程在正常器官及神经系统发育过程中至关重要。糖基化的生理过程非常复杂,需要100多个步骤,每个步骤需要相应酶的催化才能完成。每个酶的反应精确有序地对蛋白质(或脂肪)添加特定的糖分子或从蛋白质(或脂肪)去除特定的糖分子。先天性糖基化障碍的患者通常存在一种或多种糖基化酶的功能缺陷,出现异常糖基化的蛋白(或脂肪),导致功能异常的糖蛋白(或糖脂),从而出现相应的临床表现。目前为止,科学家们发现118种先天性糖基化障碍,每型均有独特的糖基化酶缺陷。大部分糖基化障碍与N-连接寡聚糖合成异常有关。人体所有细胞均需要按特定顺序合成寡聚糖,形成不同糖链与蛋白结合。寡聚糖对蛋白稳定性及细胞间信号传递所必须,寡聚糖合成异常可能导致不同器官功能的异常。因糖基化过程非常复杂,今后也会不断发现新的先天性糖基化障碍。二、先天性糖基化障碍分型及命名先天性糖基化障碍(CDG)是一组罕见的遗传代谢病,影响复杂的糖基化酶代谢过程。先天性糖基化障碍(CDG)根据生物化学通路缺陷发生的部位分为I型及II型,另根据发现疾病的时间顺序添加相应字母。比如,第一次发现的先天性糖基化障碍I型被命名为IA型,第二次发现的I型命名为IB型,以此类推。此外,IX型或IIX型均表示新发现的糖基化障碍,但尚未明确相关基因或酶功能的异常。先天性糖基化障碍I型(CDG-I)是合成大糖链通路相关的基因异常(导致相应的酶功能缺陷)或大糖链与蛋白结合所需的基因异常(导致相应的酶功能缺陷)。部分CDG-I由于动员小糖单元的酶功能缺陷,或添加小糖单元到蛋白质的酶功能缺陷导致。先天性糖基化障碍II型(CDG-II)是由于催化改变与蛋白结合糖链结构的酶功能缺陷导致。包括剪除多余糖分子的糖苷酶缺陷,或改变糖分子结构的糖基转移酶的功能缺陷。部分CDG-II由于转运活化的糖分子至高尔基体(细胞的糖基化工厂)的转运蛋白功能缺陷导致。与先天性糖基化障碍相关的的基因有数百种,部分CDG-II除了蛋白糖基化障碍,同时存在糖脂及糖胺聚糖等糖链合成缺陷。相关基因的严重突变均有可能导致先天性糖基化障碍。关于先天性糖基化障碍的研究进展迅速,已发现CDG-IA到CDG-IL及IIA到IIF等疾病。多数疾病只有1到2例患者,确切临床表现尚不明确。科学家们努力发现更多的糖基化障碍患者,新的临床表现及新的糖基化障碍相关疾病。既往先天性糖基化障碍(CDG)命名方式为分型方式(比如PMM2基因缺陷导致致的命名为CDG-IA型),而目前科学家制定了新的命名方式:基因名称-CDG(比如CDG-IA型目前称之为PMM2-CDG)。新的命名方式有助于认识基因与疾病的关系,让家属及科学研究人员精确跟踪病情变化。三、临床表现每一位先天性糖基化障碍患儿临床表现及疾病严重程度不同,部分症状到了一定年龄才能表现的比较重。先天性糖基化障碍影响人体多数器官系统。(一)以下是至今发现与CDG相关的临床表现(患儿可能至少会有3-4种类似表现,强调全面评估的重要性):特殊面容(前额突出、杏仁状眼睛、眼窝凹陷、眉毛高拱、耳廓偏大、鼻梁短、鼻梁塌陷、人中偏长且光滑、上唇偏薄且前凸)特殊的体表特征(乳头内陷、短指或长指畸形、指/趾重叠、指/趾弯曲、全身水肿、皮肤发红或脱皮、皮下脂肪异常分布、多毛、皮肤橘皮样改变)腹泻、消化或吸收障碍、丢失蛋白型肠病、体重生长缓慢、营养不良肝脏疾病或肝功能指标异常(白蛋白低下、转氨酶升高、肝肿大、脾肿大,肝硬化、肝纤维化、肝功能衰竭、肝囊肿、胆汁淤积症)凝血功能异常、出血、血栓形成腺体功能异常(甲状腺功能低下、高雄激素血症、女性缺乏性发育特征、女性出现男性化特征)免疫功能异常(免疫球蛋白水平下降、容易感染)神经系统异常(脑发育不良或脑萎缩、髓鞘化延迟、脑积水、假脑瘤、周围神经系统病变)肌张力低下、抽搐、发作性肌张力升高、脑中风样发作运动发育落后、智力障碍小脑发育不良或小脑萎缩(大脑影像学检查发现)共济失调(平衡或运动协调能力差)发音不良,语言落后眼部异常(斜视、内斜视、白内障、角膜混浊、虹膜缺损、眼球震颤、视网膜病变)、视网膜色素变性(色素性视网膜炎)脊柱侧弯、脊柱后凸、关节挛缩或屈曲心脏异常(心包积液、法洛四联症、永存动脉干)(二)部分先天性糖基化障碍出现的特殊临床表现先天性糖基化障碍IIC型(SLC35C1-CDG)表现为严重的营养不良、发育落后、小脑畸形、肌张力低下、特殊面容、反复细菌感染及白细胞升高。先天性糖基化障碍IB型(MPI-CDG)则表现不同,患儿神经系统发育正常,但可能会出现肠道吸收及消化功能严重缺陷,丢失蛋白性肠病、肝功能异常,白蛋白水平低,低血糖、凝血功能障碍,血栓形成。先天性糖基化障碍IT型(PGM1-CDG)表现为肝功能异常、悬雍垂裂、低血糖、乳酸升高、营养不良、矮小、扩张性心肌病、骨骼肌异常(运动不耐受,横纹肌溶解),性激素异常。四、诊断自从1980年首次报道并认识先天性糖基化障碍这组疾病,对此病的认识不断加深,早期诊断及分类方法得到进一步改进。任何不明原因的发育落后、营养不良、脑中风样发作、抽搐、小脑功能异常及肝功能异常的患儿应接受检查并确诊。大部分先天性糖基化障碍可以通过简单的血液检测确定转铁蛋白糖基化程度进行初步诊断。异常糖基化的转铁蛋白是通过等电聚焦(IEF)方法或电喷雾电离质谱(ESI-MS)检测方法确定。初步诊断糖基化障碍后需要进一步检测基因、酶活性测定及功能试验等进一步确诊。虽然先天性糖基化障碍被认为是罕见病,越来越多的证据表明此病可能比想象中的要多见。全世界诊断的1200例患者可能只代表所有糖基化障碍病人冰山一角,可以说先天性糖基化障碍病人确诊率很低,实际发病率及患者数量尚不明确,每5000个人中有一例先天性糖基化障碍患者。早期的先天性糖基化障碍病人由于症状与其他遗产代谢病类似而可能被误诊。最常见的PMM2-CDG(CDG-1A)患者有时候被误诊为线粒体疾病或共济失调性脑瘫。五、先天性糖基化障碍与肝脏疾病糖蛋白合成是肝脏主要功能之一,过去20年的研究发现众多CDG出现细胞膜及分泌的糖蛋白异常影响肝脏结构及功能,导致脂肪肝、肝纤维化及胆管病变。异常折叠的糖蛋白在内质网蓄积,异常糖基化的血浆蛋白稳定性差,导致高凝或低凝状态。部分CDG病变仅限于肝脏,但多数伴有其他系统病变,包括胃肠道(蛋白丢失性肠病)、低血糖、肌张力低下、发育落后及抽搐。血清铁蛋白糖基化实验(铁蛋白等电聚焦电泳)通过简单而成本低的方法筛查CDG,所有不明原因肝病患者应筛查CDG,合并有其他系统病变的患者尤为如此。与肝脏相关的CDG大致可分为两大类,单纯表现为肝脏病变或肝脏病变为主的CDG(包括MPI-CDG、TMEM199-CDG、CCDC115-CDG,及ATP6AP1-CDG)及其他系统病变为主但合并有肝脏损害的CDG(包括PMM2-CDG、ALG1-CDG、ALG3-CDG、ALG6-CDG、ALG8-CDG、ALG9-CDG、PGM1-CDG及COG-CDG)。进一步认识及研究CDG相关肝损害不仅有助于早期诊断,早期治疗,也帮助改善CDG患者长期预后。肝脏病变是CDG常见的表现之一,可出现肝硬化及肝功能衰竭等严重病变。多数CDG报道转氨酶升高,ALT最高值往往出现在发热时或用抗癫痫药物后。肝脏合成的糖蛋白减少,尤其是与凝血功能相关的糖蛋白,包括蛋白C、抗凝血酶III(AT-III)及凝血因子(VII、IX及XI)。常见的肝脏病理改变包括脂肪肝、汇管区周围纤维化及肝细胞肿胀,而极少见到炎症表现。部分患者肝脏病理表现为胆管板发育不良及胆管扩张等典型的纤维囊性病变。肝脏电镜超微结构主要表现为类似于尼曼匹克C(NPC)型的溶酶体包涵体,可能提示NPC2蛋白糖基化异常导致胆固醇转运障碍。然而,与NPC不同,溶酶体内包涵体仅见于肝细胞,枯否细胞溶酶体正常。CDG患者血浆多重溶酶体酶水平升高,提示细胞内转运(富含甘露糖结构)及溶酶体靶标(甘露糖-6-磷酸,Man-6-P)相关糖基化异常。此外,肝细胞脂肪变性多见于汇管区周围,并且杆细胞内脂褐素(lipofuscin),提示三肽氨基肽酶(tripeptidyl peptidase)糖基化水平低下影响蛋白折叠、转运功能及稳定性。先天性糖基化障碍与多囊肝:虽然多囊肝是少见病,约25%的患者发现内质网表达的PRKCSH或SEC63基因突变。PRKCSH-CDG及SEC63-CDG均与内质网糖基化过程相关且引起多囊肝病,其他内质网糖基化相关的基因突变也可以出现肝脏病变。如,两例ALG3-CDG患者出现肝肿大、胆湖形成、肝纤维化及胆管板发育不良;三例ALG9-CDG患者出现肝肿大,其中一例发现肾囊肿;ALG12-CDG患者虽然没有发现肝肿大,但转氨酶升高;ALG6-CDG患者可出现肝肿大,其中三例肝活检发现肝细胞内异常溶酶体包涵体,但未发现胆管异常;ALG8-CDG可出现严重的转氨酶升高及肝肿大,一例肝穿患者发现肝内外胆管多发囊性扩张,随访时出现胆汁淤积及肾脏微小囊肿;一例新生儿GLS1-CDG患者出现进行性肝肿大74日龄死亡,尸检发现胆管增生扩张、胆汁淤积、脂肪变性、肝纤维化、毛细胆管及肝细胞内胆栓样改变。
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科1.4万人已读 - 精选 先天性糖基化障碍治疗
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生(一)以下先天性糖基化障碍类型可能有特定的治疗方案(多数为研究阶段的治疗方案,建议在有经验的机构临床评估、基因诊断、糖谱分析、权衡利弊、签署知情同意书后,严密观察临床表现及糖谱变化的前提下进行治疗):(1)先天性糖基化障碍IB型(MPI-CDG):患儿神经系统发育正常,但肠道吸收及消化功能严重缺陷,丢失蛋白性肠病、肝功能异常,白蛋白水平低,低血糖、凝血功能障碍。如果早期诊断,甘露糖(mannose)治疗可能明显改善腹泻症状及低血糖,治疗后白蛋白水平及凝血功能可恢复正常。一例患者反复血栓形成及凝血功能障碍的MPI-CDG患者,甘露糖治疗后未再出现血栓,凝血功能恢复正常。有学者报道2例患儿甘露糖治疗后仍未能阻断肝纤维化出现。但一例28岁甘露糖及肝素治疗后仍出现肝纤维化的女性患者,肝移植治疗后症状缓解,随访2年仍正常。治疗腹泻及肠病方面肝素治疗可替代甘露糖治疗。(2)先天性糖基化障碍IT型(PGM1-CDG):表现为肝功能异常、悬雍垂裂、低血糖、乳酸升高、营养不良、矮小、扩张性心肌病、骨骼肌异常(运动不耐受,横纹肌溶解),性激素异常。口服半乳糖或乳糖治疗后肝功能可好转,糖基化异常得以改善,性激素水平上升、未再出现横纹肌溶解,脂肪肝及心功能指标未出现继续加重的情况。(3)先天性糖基化障碍IIc型(SLC35C1-CDG):又称GDP-岩藻糖跨膜转运蛋白缺乏症,导致GDP-岩藻糖无法转运至高尔基体。表现为严重的营养不良、发育落后、小脑畸形、肌张力低下、特殊面容、反复细菌感染及白细胞升高。细胞及动物模型添加岩藻糖后不仅改善糖基化异常,也能纠正生化及功能学指标。患者岩藻糖(fucose)治疗后改善生化异常及临床症状,白细胞计数升到正常水平,反复感染发作得以停止,改善糖基化异常指标,运动发育功能的得到了部分改善。SLC35C1-CDG患者因不能表达α1,2-岩藻糖华的H抗原,故会有孟买血型。部分患者岩藻糖治疗后出现H抗原表达,虽然尚未报道自发溶血,应警惕岩藻糖治疗后可能会出现自身免疫反应。也有部分患者岩藻糖治疗无效,可能与基因突变类型、病情严重程度等因素有关。(4)先天性糖基化障碍Ia型(PMM2-CDG):又称磷酸甘露糖酶2缺乏症。虽然有个别研究发现患者皮肤成纤维细胞培养液添加甘露糖可以改善糖基化异常,但其他体外或体内研究均未得到类似的结果。PMM2-CDG患者口服甘露糖后吸收良好,血糖升高,未引起任何肝脏或肾脏等毒性表现,但也未发现明显的临床或生化指标的改善。甘露糖治疗后虽然未得到糖基化方面的改善,部分家长反映患儿精神运动功能有所好转。甘露糖治疗失败的原因可能是因为基因突变的细胞无法摄取甘露糖,有报道称二甲双胍体外试验可促进PMM2基因突变的细胞吸收甘露糖并改善糖基化异常,也有公司在研发人体可吸收的甘露糖-1-磷酸,将来有望能帮助PMM2糖基化患者病情。(5)先天性糖基化障碍IIm型(SLC35A2-CDG):由于UDP-半乳糖转位蛋白异常导致UDP-半乳糖无法转运至高尔基体出现糖基化异常。人体研究每日每公斤体重1g的半乳糖治疗6个月后患者铁蛋白糖基化异常恢复正常。有趣的是,体外细胞实验中添加半乳糖未能改善糖基化异常,可能是因为体内和体外环境差异所致。(6)先天性糖基化障碍Iz型(CAD-CDG):又称尿苷(尿嘧啶核苷)单磷酸合成酶缺乏症或遗传性乳清酸尿症。患者补充尿苷(Uridine)治疗(每日每公斤体重100mg,分4次)后有治疗效果。体外细胞研究中,添加尿苷后糖基化异常明显改善。有人报道尿苷治疗2例CAD-CDG患者,临床表现显著改善,未再抽搐发作,感知和运动发育明显好转,对外界反映明显好转,交流能力进步,生化指标也恢复正常。(7)先天性糖基化障碍IIf型(SLC35A1-CDG):是转运胞苷一磷酸-唾液酸至高尔基体的蛋白功能缺陷导致的唾液酸糖基化障碍。体外细胞研究添加唾液酸、乙酰甘露糖胺或糖基化蛋白胎球蛋白措施均未改善患者体内提取的细胞糖基化异常。(8)GNE-先天性糖基化障碍(GNE-CDG):又称尿苷二磷酸-N-乙酰葡糖胺-2-差向异构酶/N-乙酰甘露糖胺激酶缺乏症,GNE-肌病或包涵体肌病,导致唾液酸合成障碍。表现为骨骼肌进行性肌无力。补充乙酰甘露糖胺(ManNAc)及D-甘露糖胺(ManN)等唾液酸前体在体外实验中可以替代GNE功能缺陷。多数GNE-CDG患者成人期发病,成人老鼠模型研究中补充ManNAc及ManN能够改善肾脏及肌肉的唾液酸化障碍,但只有ManNAc能改善蛋白尿。Ac4ManNAc效果更好。6′-唾液乳糖代谢缓慢,更能被细胞吸收,明显改善老鼠模型肌肉功能及唾液酸化异常,目前已有人体临床试验在进行中。(9)NANS-先天性糖基化障碍(NANS-CDG):又称CMP-N-乙酰神经氨酸合成酶缺乏症,影响唾液酸生物合成通路。已报道9例患儿,表现为婴儿期起病的严重发育落后,特殊面容(前额凸出、鼻梁塌陷、鼻尖饱满、嘴唇丰满)及骨骼发育不良(矮小、腕关节过早骨化、扁椎骨、干骺端纵向条纹及骨骺偏小)。体液检查N-乙酰-D-甘露糖胺水平升高,但血浆蛋白唾液酸化程度正常,故Tf-IEF正常。斑马鱼疾病模型中受精24小时后用唾液酸治疗可改善骨骼病变,但胎儿期后治疗有效性不清楚。有研究者已提交唾液酸缓释药物治疗NANS-CDG患者临床试验申请,但尚未应用于人体试验。(10)PGM3-先天性糖基化障碍(PGM3-CDG):又称葡糖磷酸变位酶3缺乏症,影响UDP-N-乙酰葡萄糖胺合成。临床表现多样,包括过敏性疾病、自身免疫性疾病、皮肤病、感染性疾病、神经系统病变、骨骼异常、肺部疾患及肾脏疾病。多数患者化验发现白细胞减少及IgE升高、部分患者仅表现为免疫缺陷。Tf及apo C-III IEF正常。患者预防性抗生素应用可能减少感染,但补充免疫球蛋白无效。体外细胞研究发现补充N-乙酰葡萄糖胺(GlcNAc)后UDP-N-乙酰葡萄糖胺合成恢复正常,但人体试验未能取得疗效,故临床研究已被取消。(11)先天性糖基化障碍Ik型(ALG1-CDG):又称壳二糖二磷酸长醇β-甘露糖苷转移酶缺乏症,导致LLOs甘露糖基化异常。表现为神经系统、眼部、心脏、肝脏、肾脏、性腺及胰腺分泌胰岛素的β细胞异常。ALG1-CDG患者体内提取的细胞中添加甘露糖可改善糖基化异常,提示人体试验可能有效。(12)先天性糖基化障碍Is型(ALG13-CDG):ALG13基因突变引起的N-糖基化障碍,导致神经系统、眼部及肝脏异常。携带p.E463G突变的病人提取成纤维细胞后,体外培养液添加D-半乳糖后疾病标记物ICAM-1水平升到正常,提示半乳糖治疗可能对ALG13-CDG患者治疗作用。(13)MAGT1-先天性糖基化障碍(MAGT1-CDG):又称镁转运蛋白1缺乏症,影响N-糖基化导致XMEN综合征(X-联锁免疫缺陷病,镁离子缺乏症,EB病毒感染及肿瘤)。体内及体外研究补充镁离子取得了令人鼓舞的效果,有报道1例患者口服补充镁,但未报道随访结果。(14)PIGA-先天性糖基化障碍(PIGA-CDG):又称磷脂酰肌醇N-乙酰葡糖胺转移酶A亚单位缺乏症,导致MCAHS2综合征(多重先天畸形-肌张力低下-惊厥综合征2)。两兄弟抗惊厥药物治疗无效,但生酮饮食治疗后病情明显改善,未再出现抽搐,发育状况进步。需要注意的是,生酮饮食可能会导致或加重低血糖。(15)PIGM-先天性糖基化障碍(PIGM-CDG):PIGM基因启动子区突变后糖基磷脂酰肌醇α-1,4-甘露糖转移酶I功能缺陷,导致组蛋白乙酰化障碍,基因转录及表达异常。有学者报道体外或体内进行丁酸钠治疗均促进乙酰化及PIGM基因转录,患者惊厥得到控制,神经系统功能得到改善。这些数据也表明组蛋白去乙酰化酶抑制剂也可能对PIGM-CDG有治疗作用。(16)PIGO-先天性糖基化障碍(PIGO-CDG):又称糖基磷脂酰肌醇磷酸乙醇胺转移酶3缺乏症,表现为高磷酸酶症、智力障碍、顽固性惊厥。患者口服维生素B6(20mg/kg)惊厥停止发作,可能因为患者维生素B6缺乏导致大脑GABA合成障碍。(17)先天性糖基化障碍IIk(TMEM165-CDG):导致锰离子(Mn2+)无法转运至高尔基体,患者可表现为精神运动发育异常、生长落后或矮小症、特殊面容、肌张力低下、眼部异常、获得性小头畸形、肝肿大及骨骼发育异常。体外试验补充锰离子可使糖基化异常得到改善。两例携带同样纯合突变而且Mn2+水平正常的患者补充半乳糖(1g/Kg*天)后生化及N-糖基化指标均有改善。然而,R126H及E108G等不同突变对Mn2+敏感性也不同,提示基因突变可能与疗效有关。(18)先天性糖基化障碍IIo(CCDC115-CDG):影响V-ATPase功能,减少细胞内铁离子水平及高尔基体稳态。表现为明显的肝功能异常、高胆固醇血症、精神运动障碍及肌张力低下。体外细胞模型补充柠檬酸铁改善相关的酶活性。如患者提取的细胞试验也得到类似效果,补铁治疗可能对CCDC115-CDG患者有效。(19)先天性糖基化障碍IIp(TMEM199-CDG):也影响V-ATPase功能,TMEM199 –CDG发病机制可能与CCDC115-CDG类似。患者表现为轻度肝功能异常,也应进一步研究补铁治疗对TMEM199-CDG患者疗效。(20)先天性糖基化障碍IIn(SLC39A8-CDG):此病引起锰(Mn2+)、锌(Zn2+)及镉(Cd2+)等离子代谢障碍导致糖基化异常。表现为智力障碍、发育落后、肌张力低下、斜视、小脑萎缩、矮小症、血Mn及Zn水平下降。有人报道补充半乳糖、尿苷(促进UDP-半乳糖合成)、Mn2+(患者血中无法检测到锰元素)2周治疗后糖基化异常改善。继续单用Mn2+治疗12个月发现生化指标及临床表现明显好转。虽然半乳糖治疗可以纠正糖基化异常,但无法纠正Mn2+缺乏引起的酶功能异常。(21)ISPD-先天性糖基化障碍(ISPD-CDG):为合成CDP-核糖醇的酶缺陷导致O-糖基化障碍,表现为先天性肌肉萎缩、肌张力低下、神经系统及眼部异常,患者一般不能活过儿童早期。体外研究细胞培养液添加核糖醇或核糖醇代谢物后明显增加糖基化。患者提取的细胞经过体外治疗后糖基化功能恢复,细胞形态恢复正常,提示核糖醇可以作为潜在的治疗药物。(二)研究中的其他治疗策略(1)药物分子伴侣(Pharmacological Chaperones),可能对导致蛋白折叠异常的错义突变,可能改善蛋白折叠,稳定蛋白功能。多数PMM2-CDG患者基因突变为错义突变,影响蛋白折叠,将来有望药物分子伴侣治疗改善患者病情。(2)反义治疗(Antisense Therapy):可能对剪切位点突变及导致新剪切位点或外显子的内含子区突变有效。已有体外细胞模型研究反义治疗对PMM2-CDG及TMEM165-CDG的影响。(3)基因治疗(Gene Therapy):糖基化障碍均为单基因突变引起的疾病,基因治疗后将突变的基因替换为正常的基因可能纠正病因。GNE-CDG患者提取的肌肉细胞研究已成功表达正常的转基因,动物实验中也证实疗效,治疗10周后骨骼肌、肝脏、肾脏、心脏及脾脏GNE表达增加。(4)器官或细胞移植(Transplantation):已有肝移植(MPI-CDG、CCDC115-CDG及ATP6VAP1-CDG),心脏移植(DOLK-CDG及PGM1-CDG)及造血干细胞移植(PGM3-CDG及MAGT1-CDG)的报道。(三)以下治疗方法适用于IB型之外的所有先天性糖基化障碍患者:(1)营养不良:绝大数先天性糖基化障碍婴儿及儿童均有明显的营养不良,体重计身高测量指标均不达标。保证充足营养的前提下可以母乳或普通奶粉喂养,但婴儿早期深度水解或氨基酸奶粉喂养似乎能够改善营养状况。先天性糖基化障碍患者不需要限制饮食,可以耐受碳水化合物、脂肪及蛋白质等所有营养物质。应根据患儿口腔运动协调能力及吞咽功能选择喂养方法并决定是否添加辅食。部分患儿需要通过胃管或胃造瘘管喂养,随着口腔运动协调能力及吞咽功能的提高,可以逐渐过渡至经口喂养。(2)口腔运动协调能力障碍及持续呕吐:多数先天性糖基化障碍患儿存在不同程度的吸吮及吞咽协调能力异常,导致喂养困难及体重增长缓慢,部分孩子持续溢奶或频繁呕吐,家长十分焦虑。吃奶或饮食后保持直立的姿势、添加稠厚的辅食、使用奶粉增稠剂及口服抑制胃酸的药物等方法均可减少呕吐。有时需要寻求消化科医生及营养师指导帮助。即便孩子已经有胃造瘘管喂养,应鼓励孩子经口喂养,但前提是呛奶或吸入食物的风险很低。患儿应持续接受口腔运动训练及发音训练等治疗,这样不仅能够保证顺利过渡至经口喂养,还能帮助改善语言能力。(3)发育落后:先天性糖基化障碍患儿家长通常在孩子满四个月左右开始感觉到发育落后。此时建议儿保科及康复科进行发育评估,必要时早期接受康复治疗、运动训练及语言治疗。随着孩子长大,先天性糖基化障碍患儿的发育与正常孩子差距会越来越加大,家长需要持续评估发育状况,康复治疗方案也需要相应调整。(4)肝功能异常:多数先天性糖基化障碍患儿一岁以内既有肝功能异常(转氨酶升高)。有时谷丙转氨酶(ALT)及谷草转氨酶(AST)可能会升高至1000-1500 IU/L后逐渐下降。先天性糖基化障碍IA型患儿转氨酶水平会逐渐下降,通常情况下3-5岁以后降至正常而且不再升高。部分患儿出现肝肿大、脂肪肝、肝纤维化、脾脏肿大等表现。严重的肝功能损伤、肝纤维化、脾肿大、肝肿大等情况均可能需要肝穿刺活检明确肝脏病变性质及严重程度。也有罕见肝功能衰竭的先天性糖基化障碍患儿。(5)凝血功能障碍:许多先天性糖基化障碍患者凝血因子水平偏低,但极少影响日常生活。然而,需要手术治疗时应该重视凝血功能障碍,应在外科医生及血液科医生指导下评估凝血功能及凝血因子水平,必要时输注新鲜血浆可以纠正凝血因子缺乏并且改善出血症状。部分肝功能异常的先天性糖基化障碍患者因维生素K1缺乏或肝脏合成凝血功能不足导致凝血功能异常,补充维生素K1,诊治肝脏疾病可能使凝血功能好转或恢复正常。先天性糖基化障碍1A型患儿1岁以内极少需要住院或手术治疗,而其他类型患儿满1岁之前可能需要数次寻找医生帮助。(6)斜视:儿童眼科医生早期干预治疗保持正常视力是至关重要的措施。很多先天性糖基化患儿存在内斜视,但手术治疗可成功纠正。部分患儿仅需要遮眼治疗或佩戴眼镜。(7)心包积液:许多先天性糖基化障碍1A型患儿存在心包积液,但不会出现其他不适并且会逐渐吸收。必要时在儿童心内科医生指导下检查心脏彩超评估并且随访。(8)甲状腺功能减退:许多先天性糖基化障碍患儿促甲状腺激素偏高,游离T4水平偏低,需要在儿童内分泌医生指导下接受评估,必要时甲状腺激素替代治疗。(9)抽搐:先天性糖基化障碍1A型患儿1-2岁左右可能会出现抽搐症状,但可以通过药物治疗控制抽搐发作。部分ID型及IH型患儿可能会出现顽固性难以控制的抽搐发作。需要在儿童神经科医生指导下治疗随访。(10)脑中风样发作:先天性糖基化障碍患儿4岁或更大时可能会出现短暂性神经功能丧失或脑中风样发作。部分家长反映头部外伤、脱水或发热时更容易出现类似发作。发作时可补液、保持血糖稳定、止惊等对症处理,恢复期需要康复治疗改善神经系统功能。通常1周左右可完全恢复到发作前的状态,少数情况下需要数月的康复治疗。(11)骨骼发育异常:先天性糖基化患儿到了青春期或成人期可能会出现胸廓发育不良、脊柱侧弯及脊柱后凸。可根据实际情况进行骨科手术、康复理疗、订制轮椅,更重要的是持续家庭康复治疗。既往通过手术治疗纠正脊柱弯曲的儿童及成人患者手术成功率并不是百分十百。(12)独立生活能力缺乏:随着孩子长大,家长需要考虑培养独立生活能力。学龄期需要强化生活能力技巧及职业能力方面的训练。应鼓励患儿独立完成日常生活任务,培养生活自理的能力。必要时在康复医师指导下使用辅助工具及资源。
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科1万人已读 - 精选 先天性糖基化障碍与肝脏疾病
复旦大学附属儿科医院肝病科库尔班江阿布都西库尔医生,王建设教授此文于2019年8月发表在“临床肝胆病杂志”摘要:先天性糖基化障碍(CDG)是近年来快速增长的一组遗传代谢性疾病,由蛋白或脂肪的异常糖基化所致。随着二代测序技术的问世和普及、人类在不断发现新的糖基化相关基因突变导致的疾病。糖蛋白及糖脂合成是肝脏主要功能之一,众多的CDG影响肝胆结构及功能,可导致脂肪肝、肝纤维化及胆管板发育异常。报道了CDG及相关肝脏病变的发病机制、诊断及治疗等方面的新进展。关键词:先天性糖基化病;诊断;糖组学中图分类号:R575.29文献标志码:A文章编号:Congenital Disorders of Glycosylation and Liver DiseasesABUDUXIKUER Kuerbanjiang, WANG Jian-She.(Department of Hepatology, Children’s Hospital of Fudan University, Shanghai 201102)Abstract:Congenital disorders of glycosylation (CDG) is a group genetic disorders that lead to abnormal glycosylation of proteins and lipids, and number of CDGs are increasing rapidly in recent years. With the advent and popularization of next generation sequencing technology, the number of new disorders related to glycosylation is increasing. One of the major roles of the liver is synthesis of glycoproteins and glycolipids, and many CDGs affect hepatobiliary structure or function leading to steatosis, fibrosis, and ductal plate malformation. In this report, we summarize latest advances in diagnosis and treatment of CDGs and related liver disorders.Key words:congenital disorders of glycosylation; diagnosis; glycomics1概述先天性糖基化障碍(congenital disorders of glycosylation,CDG)是近年来快速增长的一组遗传代谢性疾病[1-2],由蛋白或脂肪的异常糖基化所致[3]。因表现多样,多器官系统功能异常,临床诊断往往困难。随着二代测序技术的问世和普及、哺乳动物细胞模型的成熟及糖组学检测技术的进步,人类在不断发现新的糖基化相关基因突变导致的疾病[4-7]。1.1糖基化过程糖基化是通过酶的催化,糖和蛋白(或脂肪)结合形成糖蛋白(或糖脂)的生物化学过程。糖与蛋白(或脂肪)结合后使蛋白质(或脂肪)与相应器官组织链接,保证其功能正常运行。糖基化过程在正常器官及神经系统发育过程中至关重要[8]。糖基化的生理过程非常复杂,需要100多个步骤,每个步骤需要相应酶的催化才能完成。每个酶的反应精确有序地对蛋白质(或脂肪)添加特定的糖分子或从蛋白质(或脂肪)去除特定的糖分子。CDG的患者通常存在一种或多种糖基化酶的功能缺陷,出现异常糖基化的蛋白(或脂肪),导致糖蛋白(或糖脂)功能异常,从而出现相应的临床表现[9]。目前为止,科学家们发现130多种CDG,每型均有独特的糖基化酶缺陷。大部分糖基化障碍与蛋白质和天门冬酰胺结合的N-连接寡聚糖合成异常有关,被称为N-连糖基化障碍(图1)。人体所有细胞均需要按特定顺序合成寡聚糖,形成不同糖链与蛋白结合。寡聚糖对蛋白稳定性及细胞间信号传递起非常重要的作用,是必不可少,寡聚糖合成异常可能导致不同器官功能的异常。因糖基化过程非常复杂,今后也会不断发现新的CDG。此外还有O-连糖基化障碍、N和O-连联合糖基化障碍、脂肪糖基化障碍及糖基磷脂酰肌醇(glycosylphosphatidylinosi-tol, GPI)锚生物合成障碍。O-连糖基化过程在高尔基体进行,逐步在糖基转移酶的作用下向蛋白中丝氨酸、苏氨酸及羟基赖氨酸添加糖链。GPI锚是内质网内合成,高尔基体内修饰的糖脂。GPI锚生物合成完成后在细胞膜定位并与数百种细胞膜蛋白结合行使众多细胞功能[10-13]。图1N-连接糖基化通路及相关基因[4]动物模型已用于CDG研究,包括无脊椎动物(果蝇及秀丽隐杆线虫)及脊椎动物(非洲爪蟾、泰和鸡、斑马鱼、小家鼠及大鼠)。小鼠由于获取容易、体积小、繁殖速度快、饲养及研究成本低以及人类基因同源相似度高等特点,已成为研究人类遗传性疾病理想的动物模型。已建立的小鼠CDG模型中,PMM2-CDG (CDG-Ia)、MPI-CDG (CDG-Ib)、DPAGT1-CDG (CDG-Ij)及SRD5A3-CDG (CDG-Iq)等内质网糖基化障碍相关基因敲除小鼠模型均在胚胎发育早期或中期死亡,提示内质网糖基化过程异常时胚胎发育晚期组织分化及器官形成过程受到严重影响。而MGAT2-CDG (CDG-IIa)、SLC35C1-CDG (CDG-IIc)及B4GALT1-CDG (CDG-IId)等高尔基体糖基化障碍相关基因敲除小鼠模型胚胎发育后出生,可短期内存活,甚至表现出类似于人类患者的表型,提示无论高尔基体上N-乙酰葡糖胺、岩藻糖或半乳糖均不是胚胎发育所必须的碳水化合物。新一代小鼠模型可利用亚等位基因敲除来避免胚胎期死亡,并且同时敲除N-糖基化通路的不同基因进一步明确糖基化障碍对胎儿及出生后不同时期发育的影响[14-15]。1.2 CDG命名及遗传模式CDG是一组罕见的遗传代谢病,影响复杂的糖基化酶代谢过程。既往CDG根据生物化学通路缺陷发生的部位分为I型及II型,另根据发现疾病的时间顺序添加相应字母(比如PMM2基因缺陷导致的命名为CDG-Ia型),而随着基因诊断技术的普及,科学家们制订了新的命名方式:基因名称-CDG(比如CDG-Ia型目前称之为PMM2-CDG)。新的命名方式有助于认识基因与疾病的关系,让家属及科学研究人员精确跟踪病情变化。绝大多数CDG遵循常染色体隐性遗传模式,仅有少数CDG遵循常染色体显性(N-连CDG:GANAB-CDG及PRKCSH-CDG;O-l连CDG:EXT1/EXT2-CDG, POFUT1-CDG及POGLUT1-CDG)或X-连锁遗传模式(ALG13-CDG、SSR4-CDG、PIGA-CDG、SLC35A2-CDG及ATP6AP1-CDG)。大部分常染色体显性及X-连锁遗传的CDG患者由相关基因自发(de novo)突变导致[16-17]。1.3 CDG临床表现[1,7,12-13,18-19]每一种CDG的临床表现及疾病严重程度可有不同,但多影响人体多个器官系统,部分器官的症状到了一定年龄才表现出来。1.3.1多种CDG可能出现的共有临床表现(患儿可能至少会有3~4种类似表现,强调全面评估的重要性)特殊面容(前额突出、杏仁状眼睛、眼窝凹陷、眉毛高拱、耳廓偏大/下移/后旋、鼻梁短、鼻梁塌陷、人中偏长且光滑、嘴唇丰厚、上唇偏薄且前凸)、特殊的体表特征(乳头内陷、短指或长指畸形、指/趾重叠、指/趾弯曲、全身水肿、皮肤发红或脱皮、皮下脂肪异常分布、多毛、皮肤橘皮样改变)、腹泻或营养不良、肝功能指标异常(白蛋白低下、转氨酶升高、胆汁淤积、肝衰竭、肝肿大、脾肿大、肝硬化、肝纤维化)、凝血功能异常、腺体功能异常、免疫功能异常、神经系统异常、运动发育落后、智力障碍、共济失调、语言落后、眼部异常、脊柱或关节病变及心脏异常。1.3.2部分CDG的特殊临床表现SLC35C1-CDG(IIc型)表现为严重的营养不良、发育落后、小脑畸形、肌张力低下、特殊面容、反复细菌感染及白细胞升高。MPI-CDG(Ib型)则表现不同,患儿神经系统发育正常,但可能会出现肠道吸收及消化功能严重缺陷,蛋白丢失性肠病、肝功能异常,白蛋白低,低血糖、凝血功能障碍及血栓形成。PGM1-CDG(It型)表现为肝功能异常、悬雍垂裂、低血糖、乳酸升高、营养不良、矮小、扩张性心肌病、骨骼肌异常(运动不耐受,横纹肌溶解),性激素异常。1.4 CDG诊断由于CDG相关基因(130多个基因)及表型(177种独特表型)众多、不同CDG可出现相同表型,而同一CDG表型可截然不同、缺乏简单快速筛查方法及医务人员对CDG认识不足等众多原因,CDG诊断面临巨大的挑战。大部分CDG可以通过血液检测初步筛查。CDG筛查第一步应为血清转铁蛋白(transferrin,Tf)等电聚焦电泳(isoelectrofocusing,IEF)及电喷雾电离质谱(ESI-MS)检测方法确定检查异常糖基化的Tf,但毛细管区带电泳及高效液相色谱检查方法速度更快且省力。然而,Tf-IEF只能检测N-糖基化障碍,无法检测O-糖基化引起的脂肪糖基化障碍及GPI锚合成障碍。约50%的CDG无法通过Tf-IEF方法检测,而且目前缺乏通用于所有CDG的检测方法。少数情况下临床表现及IEF/质谱等方法考虑特定CDG时单基因测序及相关酶活性测定可帮助确诊,而多数情况下临床表现及糖谱检查特异性差,需要包括所有已知CDG相关基因的基因包(panel)甚至全外显子组等高通量测序技术帮助诊断[20-22]。1.5 CDG治疗[23-25]1.5.1 CDG特定类型的特异治疗方案(1)MPI-CDG(Ib型)患儿表现为神经系统发育正常,但肠道吸收及消化功能严重缺陷,蛋白丢失性肠病、肝功能异常,白蛋白低,低血糖、凝血功能障碍。如果早期诊断,甘露糖治疗可能明显改善腹泻症状及低血糖,治疗后白蛋白水平及凝血功能可恢复正常。1例反复血栓形成及凝血功能障碍的MPI-CDG患者,甘露糖治疗后未再出现血栓,凝血功能恢复正常。有学者报道2例患儿甘露糖治疗后仍未能阻断肝纤维化出现。但1例28岁甘露糖及肝素治疗后仍出现肝纤维化的女性患者,肝移植治疗后症状缓解,随访2年仍正常。治疗腹泻及肠病方面肝素治疗可替代甘露糖治疗。(2)PGM1-CDG(It型)表现为肝功能异常、悬雍垂裂、低血糖、乳酸升高、营养不良、矮小、扩张性心肌病、骨骼肌异常(运动不耐受,横纹肌溶解),性激素异常。口服半乳糖或乳糖治疗后肝功能可好转,糖基化异常得以改善,性激素水平上升、未再出现横纹肌溶解,脂肪肝及心功能指标未出现继续加重的情况。(3)SLC35C1-CDG(IIc型)表现为严重的营养不良、发育落后、小脑畸形、肌张力低下、特殊面容、反复细菌感染及白细胞升高。患者使用岩藻糖治疗可能会控制反复感染,改善糖基化异常指标。1.5.2体外或动物实验提示可能有效的治疗措施CAD-CDG进行尿苷治疗、SLC35A1-CDG进行唾液酸治疗、GNE-CDG补充乙酰甘露糖胺及D-甘露糖胺等唾液酸前体、NANS-CDG进行唾液酸缓释药物治疗、PGM3-CDG补充N-乙酰葡萄糖胺、ALG1-CDG进行甘露糖治疗、ALG13-CDG进行D-半乳糖治疗、MAGT1-CDG补充镁离子、PIGA-CDG生酮饮食、PIGM-CDG进行丁酸钠治疗、PIGO-CDG口服维生素B6、TMEM165-CDG补充锰离子或半乳糖、CCDC115-CDG补充柠檬酸铁、TMEM199-CDG补铁治疗、SLC39A8-CDG补充半乳糖级尿苷等促进UDP-半乳糖合成和Mn2+治疗、ISPD-CDG核糖醇或核糖醇代谢物治疗。1.5.3对症、支持治疗营养不良、口腔运动协调能力障碍及持续呕吐、发育落后、肝功能异常、凝血功能障碍、斜视、心包积液、甲状腺功能减退、抽搐、脑中风样发作(发作时可补液)、骨骼发育异常及独立生活能力缺乏应该对症处理。2 CDG与肝脏糖蛋白及糖脂合成是肝脏主要功能之一,过去20年的研究发现众多CDG出现细胞膜及分泌的糖蛋白异常影响肝脏结构及功能,导致脂肪肝、肝纤维化及胆管病变。异常折叠的糖蛋白在内质网蓄积影响细胞功能,异常糖基化的血浆蛋白及凝血因子稳定性差,导致高凝或低凝状态。部分CDG病变仅限于肝脏,但多数伴有其他系统病变,包括胃肠道(蛋白丢失性肠病)、低血糖、肌张力异常、发育落后及抽搐。血清Tf糖基化实验(Tf-IEF)通过简单而成本低的方法筛查CDG。所有不明原因肝病患者应筛查CDG,合并有其他系统病变的患者尤为如此。与肝脏相关的CDG大致可分为两大类,单纯表现为肝脏病变或肝脏病变为主的CDG(包括MPI-CDG、TMEM199-CDG、CCDC115-CDG、SLC37A4-CDG及ATP6AP1-CDG)及其他系统病变为主但合并有肝损伤的CDG(包括PMM2-CDG、ALG1-CDG、ALG3-CDG、ALG6-CDG、ALG8-CDG、ALG9-CDG、PGM1-CDG及COG-CDG),其中SLC37A4-CDG又称糖原累积病Ib或Ic型。进一步认识及研究CDG相关肝损伤不仅有助于早期诊断,早期治疗,也帮助改善CDG患者长期预后[26]。表1总结了常见与肝脏病变相关的CDG肝功能相关指标变化及肝脏病理改变,而图2总结了与肝脏相关的糖基化障碍及相关蛋白在细胞内定位。图2与肝脏相关的糖基化障碍及相关蛋白在细胞内定位红色框:仅有肝脏病变或肝脏病变为主的CDG;紫色框:合并有肝脏病变的CDG[26]肝脏病变是CDG常见的表现之一,可出现肝硬化及肝衰竭等严重病变。多数CDG患者转氨酶会升高,ALT最高值往往出现在发热时或用抗癫痫药物后。肝脏合成的糖蛋白减少,尤其是与凝血功能相关的糖蛋白,包括蛋白C、抗凝血酶III(AT-III)及凝血因子(VII、IX及XI)。常见的肝脏病理改变包括脂肪肝、汇管区周围纤维化及肝细胞肿胀,而极少见到炎症表现。部分患者肝脏病理表现为胆管板发育不良及胆管扩张等典型的纤维囊性病变。肝脏电镜超微结构主要表现为类似于尼曼匹克C型的溶酶体包涵体,可能提示NPC2蛋白糖基化异常导致胆固醇转运障碍。然而,与NPC不同,溶酶体内包涵体仅见于肝细胞,而Kupffer细胞溶酶体正常。CDG患者血浆多种溶酶体酶水平升高,提示细胞内转运(富含甘露糖结构)及溶酶体靶标(甘露糖-6-磷酸)相关糖基化异常[27]。2.1 CDG与肝脏纤维囊性病变肝脏纤维囊性病变属于纤毛病,表现包括先天性肝纤维化、肝硬化、胆管扩张、胆汁淤积症、多囊肝及胆管板发育不良[28]。虽然多囊肝是少见病,约25%的患者发现内质网表达的PRKCSH或SEC63基因突变。SEC63作为转位子复合体一部分参与蛋白质在内质网内外转运,转位子复合体可将新合成的蛋白转运至粗面内质网内,也可通过内质网相关降解途径将未折叠的异常蛋白从内质网转运至胞浆内。PRKCSH基因编码葡萄糖苷酶II(又称肝囊肿蛋白hepatocystin)β亚单位,负责从蛋白质上Glc3Man9GlcNAc2 N-聚糖分裂两个葡萄糖分子。肝囊肿蛋白通过调控钙联蛋白/钙网蛋白直接参蛋白折叠过程。近期研究[29]发现内质网N-糖基化相关基因突变导致的CDG出现多囊肝病,囊上皮细胞肝囊肿蛋白表达缺失,提示糖基化障碍和多囊肝病存在共同发病机制。PRKCSH及SEC63均与内质网糖基化过程相关且引起多囊肝病,其他内质网糖基化相关的基因突变也可以出现肝脏病变。如,2例ALG3-CDG患者出现肝肿大、胆湖形成、肝纤维化及胆管板发育不良;ALG6-CDG患者可出现肝肿大,其中3例肝活组织检查发现肝细胞内异常溶酶体包涵体,但未发现胆管异常;ALG8-CDG可出现严重的转氨酶升高及肝肿大,1例肝穿患者发现肝内外胆管多发囊性扩张,随访时出现胆汁淤积及肾脏微小囊肿;3例ALG9-CDG患者出现肝肿大,其中1例发现肾囊肿;ALG12-CDG患者虽然没有发现肝肿大,但转氨酶升高;1例新生儿GLS1-CDG患者出现进行性肝肿大74日龄死亡,尸检发现胆管增生扩张、胆汁淤积、脂肪变性、肝纤维化、毛细胆管及肝细胞内胆栓样改变[29]。治疗方面应针对病因治疗改善糖基化为主,熊去氧胆酸可能对胆汁淤积症、胆管扩张、胆管板发育不良及肝囊肿有效,有望延缓或阻断肝脏病变进展。此外应监测脂溶性维生素水平,必要时补充维生素A、D、E及K1等。严重的肝囊肿及肝纤维化可能需要外科手术干预或肝移植治疗。图3引起肝脏纤维囊性病变的CDG及其发病机制[29]2.2 CDG与脂肪肝PMM2-CDG、MPI-CDG、ALG1-CDG、ALG3-CDG、ALG6-CDG、ALG8-CDG、ALG11-CDG、MOGS-CDG、COG6-CDG、CCDC115-CDG、ATP6AP1-CDG、MPDU1-CD、RFT1-CDG及PGM1-CDG等CDG均可出现不同程度的脂肪肝。CDG引起脂肪肝的发病机制尚不清楚。肝细胞脂肪变性多见于汇管区周围,并且肝细胞内脂褐素沉积,提示三肽氨基肽酶糖基化水平低下影响蛋白折叠、转运及稳定性。当然脂肪肝也可能与CDG导致调控能量及脂肪代谢通路的相关蛋白糖基化异常有关[27,30]。脂肪肝根本治疗措施仍为针对病因治疗并改善糖基化异常。此外维持血糖稳定、改善能量代谢、纠正脂肪代谢紊乱等措施可能改善肝脏脂肪病变。小结随着基因诊断技术的普及,不断发现更多CDG类型及患者。CDG与肝脏密切相关,不仅成为肝病病因之一,也为部分疑难肝病发病机制提供了有力依据。随着CDG研究深入,糖基化异常有望成为疑难肝病新的诊断措施和治疗靶点。参考文献1.SCOTT K, GADOMSKI T, KOZICZ T, et al. Congenital disorders of glycosylation: New defects and still counting[J]. J Inherit Metab Dis, 2014, 37(4): 609-617.2.JAEKEN J. Congenital disorders of glycosylation (CDG): It's (nearly) all in it![J]. J Inherit Metab Dis, 2011, 34(4): 853-858.3.JAEKEN J. Congenital disorders of glycosylation[J]. Handb Clin Neurol, 2013, 113: 1737-1743.4.NG BG, FREEZE HH. Perspectives on glycosylation and its congenital disorders[J].Trends Genet, 2018, 34(6): 466-476.5.PéANNE R, de LONLAY P, FOULQUIER F, et al. Congenital disorders of glycosylation (CDG): Quo vadis?[J]. Eur J Med Genet, 2018, 61(11): 643-663.6.JAEKEN J, PéANNE R. What is new in CDG?[J]. J Inherit Metab Dis, 2017, 40(4): 569-586.7.PARKS SE, KRASNEWICH DM. Congenital disorders of N-linked glycosylation and multiple pathway overview[M/OL]//ADAM MP, ARDINGER HH, PAGON RA, et al. GeneReviews. Seattle (WA): University of Washington, Seattle.8.REILY C, STEWART TJ, RENFROW MB, et al. Glycosylation in health and disease[J]. Nat Rev Nephrol, 2019, 15(6): 346-366.9.YAREMA KJ, BERTOZZI CR. Characterizing glycosylation pathways[J]. Genome Biol, 2001, 2(5): REVIEWS0004.10.CHANG IJ, HE M, LAM CT. Congenital disorders of glycosylation[J]. Ann Transl Med, 2018, 6(24): 477.11.van TOL W, WESSELS H, LEFEBER DJ. O-glycosylation disorders pave the road for understanding the complex human O-glycosylation machinery[J]. Curr Opin Struct Biol, 2019, 56: 107-118.12.CYLWIK B, NAKLICKI M, CHROSTEK L, et al. Congenital disorders of glycosylation. Part I. Defects of protein N-glycosylation[J]. Acta Biochim Pol, 2013, 60(2): 151-161.13.CYLWIK B, LIPARTOWSKA K, CHROSTEK L, et al. Congenital disorders of glycosylation. Part II. Defects of protein O-glycosylation[J]. Acta Biochim Pol, 2013, 60(3): 361-368.14.THIEL C, KRNER C. Mouse models for congenital disorders of glycosylation[J]. J Inherit Metab Dis, 2011, 34(4): 879-889.15.STANLEY P. What have we learned from glycosyltransferase knockouts in mice?[J]. J Mol Biol, 2016, 428(16): 3166-3182.16.WOODS AG, WOODS CW, SNOW TM. Congenital disorders of glycosylation[J]. Adv Neonatal Care, 2012, 12(2): 90-95.17.THEODORE M, MORAVA E. Congenital disorders of glycosylation: Sweet news[J]. Curr Opin Pediatr, 2011, 23(6): 581-587.18.RYMEN D, JAEKEN J. Skin manifestations in CDG[J]. J Inherit Metab Dis, 2014, 37(5): 699-708.19.GORETA SS, DABELIC S, DUMIC J. Insights into complexity of congenital disorders of glycosylation[J]. Biochem Med (Zagreb), 2012, 22(2): 156-170.20.STURIALE L, BARONE R, GAROZZO D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation[J]. J Inherit Metab Dis, 2011, 34(4): 891-899.21.WADA Y. Mass spectrometry of transferrin and apolipoprotein C-III for diagnosis and screening of congenital disorder of glycosylation[J]. Glycoconj J, 2016, 33(3): 297-307.22.ABU BAKAR N, LEFEBER DJ, van SCHERPENZEEL M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation[J]. J Inherit Metab Dis, 2018, 41(3): 499-513.23.BRASIL S, PASCOAL C, FRANCISCO R, et al. CDG therapies: From bench to bedside[J]. Int J Mol Sci, 2018, 19(5). pii: E1304.24.WITTERS P, CASSIMAN D, MORAVA E. Nutritional therapies in congenital disorders of glycosylation (CDG)[J]. Nutrients, 2017, 9(11). pii: E1222.25.THIEL C, KRNER C. Therapies and therapeutic approaches in Congenital Disorders of Glycosylation[J]. Glycoconj J, 2013, 30(1): 77-84.26.MARQUES-DA-SILVA D, DOS REIS FERREIRA V, MONTICELLI M, et al. Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature[J]. J Inherit Metab Dis, 2017, 40(2): 195-207.27.EKLUND EA, FREEZE HH. Congenital disorders of glycosylation and their effects on the liver[M]//MURRAY K, LARSON A. Fibrocystic Diseases of the Liver. USA:Humana Press.28.ROCK N, MCLIN V. Liver involvement in children with ciliopathies[J]. Clin Res Hepatol Gastroenterol, 2014, 38(4): 407-414.29.JANSSEN MJ, WAANDERS E, WOUDENBERG J, et al. Congenital disorders of glycosylation in hepatology: the example of polycystic liver disease[J]. J Hepatol, 2010, 52(3): 432-440.30.HüLSMEIER AJ, TOBLER M, BURDA P, et al. Glycosylation site occupancy in health, congenital disorder of glycosylation and fatty liver disease[J]. Sci Rep, 2016, 6: 33927.本文编辑王亚南
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科6987人已读
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科6987人已读 - 精选 PMM2先天性糖基化障碍(Ia型)
直播时间:2020年08月08日10:31主讲人:库尔班江·阿布都西库尔副主任医师复旦大学附属儿科医院肝病科
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科1219人已读 - 医学科普 ALG2先天性糖化障碍(ALG2-CDG)及ALG2先天性肌无力综合征(ALG2-CMS)
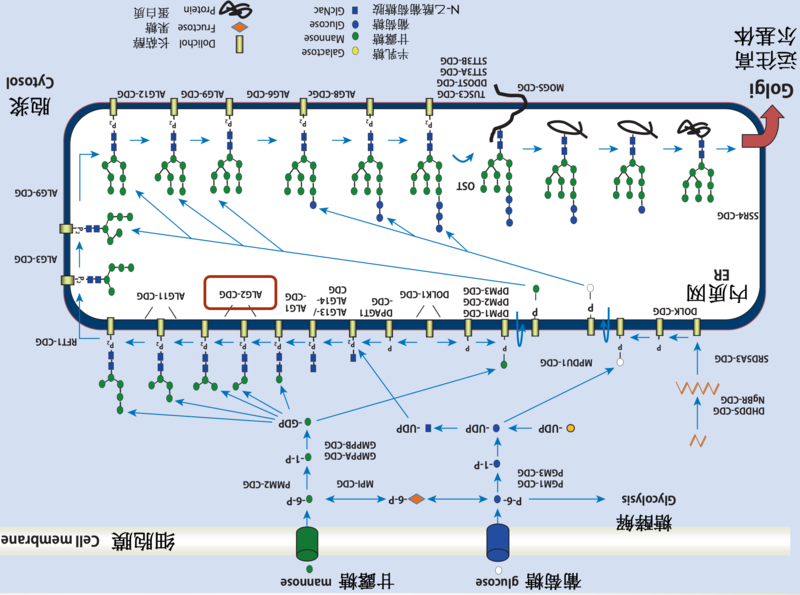
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生ALG2先天性糖化障碍(ALG2-CDG,旧称:先天性糖基化障碍Ii型)是非常罕见的糖基化障碍之一,目前为止仅报道8例,我院收治2例。ALG2基因编码负责第二及第三甘露糖糖基化过程的α-1,3甘露糖苷转移酶,ALG2基因突变影响内质网在胞浆面的糖基化从而导致蛋白糖基化障碍及功能异常(图1)。以下总结之前报道过的ALG2先天性糖化障碍的患者病情,供家长或患者参考。遗传模式ALG2-CDG为常染色体隐性遗传模式,也就是患者需要存在分别来自父母的2个ALG2基因突变,父母均为健康携带者。临床表现典型的ALG2-CDG患者一般会有包括神经系统的多器官系统异常。而轻度ALG2-CDG患者可以只出现肌无力或肌肉异常,表现为ALG2先天性肌无力综合征(ALG2-CMS)。典型的ALG2-CDG患者婴儿期发病,轻度或重度神经系统异常,伴有其他器官系统紊乱。神经系统异常:全面发育落后、智力障碍、肌力减退、肌张力低下、近端肌无力、共济失调(肌肉控制力及协调能力异常)、癫痫、婴儿痉挛症、腱反射亢进、脑电图异常(高度节律失常)眼部:视力障碍、白内障、眼球震颤及虹膜缺损。特殊面容及其他异常:小头畸形、肝肿大。ALG2-CMS患者只出现肌无力或容易乏力等肌肉异常,儿童期发病,不会出现其他器官系统异常。受累的肌肉可以是近端肌肉,远端肌肉或肢带肌肉。面部肌肉、眼肌及呼吸系的肌肉也可以出现轻度异常。既往报道过的患者2003年德国科学家Thiel等首次报道ALG2先天性糖化障碍,患者为女性,出生时除了双眼虹膜缺损没有发现其他异常。2个月龄时发现单眼白内障(手术治疗后视力仍然不是很好,不能将眼部固定到物体或其他人的脸,追视不好,经常出现眼球震颤),4个月龄起逐渐出现精神发育迟滞、惊厥(婴儿痉挛、脑电图提示高度节律失常或棘慢节律)、大脑髓鞘化延迟、肝肿大及凝血功能异常等多系统障碍。8个月龄时严重运动发育落后及严重精神发育迟滞,腱反射亢进。复查脑核磁共检查髓鞘化未见改善,脑白质明显减少。检查发现肝脏轻微肿大,心脏有轻度杂音及骶尾骨处凹陷。听力检查、常见代谢病及血液病筛查结果均正常。APTT凝血功能轻度异常,XI因子活性明显下降。转铁蛋白检查发现I型糖基化障碍。基因检查发现ALG2基因两个突变,分别为一个碱基缺失(移码突变,属于严重突变)及一个碱基替代(错义突变,替代位点决定严重程度)。患者成纤维细胞及携带基因突变的酵母菌细胞糖基化均出现异常,而不带突变的酵母菌糖基化未受到影响。患者父母均为健康的德国人,父亲家族史正常,母亲家族史有精神发育迟缓、婴儿早期死亡、抽搐、脑电图异常及偏头痛的病例。2014年沙特医生Monies等报道一个贝多因(沙漠游牧的阿拉伯民族)近亲结婚家族里面的3例ALG2-CDG患者,女性2例,男性1例,均在2-4岁发病,发病之前均可独立行走。患者III-3,目前33岁,存在近端肢带型肌营养不良及下肢肌无力。患者III-4目前18岁,下肢无力,反复跌倒。患者III-2也是18岁,存在运动发育落后及肌张力低下。患者III-4的胸锁乳突肌功能及耸肩功能正常,肌张力没有异常,但肌肉体积减少,也有肌无力的表现。坐下来站立的时候需要按照高尔氏法(Gowersmaneuver)扶着附近的凳子或桌子,停下来从地上取东西的时候出现特伦德伦堡步态(Trendelenburggait)。神经系统检查发现患者III-2及患者III-4肌力低下,下肢肌无力比上肢更为明显,近端肌力下降较远端肌力更为明显。患者III-3肌力下降跟其他两个患者类似,但上肢肌无力更为明显。所有患者均存在脊柱前凸,但患者III-2及患者III-4同时存在脊柱侧弯需要做脊柱融合手术。3例患者肌电图检查均提示肌源性损伤,患者III-3肌酸激酶轻度升高,但其他两位患者肌酸激酶水平正常。疾病进展速度不一样,患者III-3在33岁时仍然可以走路,但患者III-2及患者III-4分别在15岁及13岁时需要做轮椅。糖基化异常筛查结果提示2例轻度异常,1例正常。基因检查发现ALG2基因严重的纯合突变(c.214_224delGGGGACTGGCTdelinsAGTCCCCG/p.72_75delGDWLinsSPR)。肌肉活检提示线粒体障碍引起的肌肉损伤及神经肌肉连接处异常。2013年Cossins等报道5例ALG2-CDG患儿。患者3、4、5及患者6均为沙特表兄妹近亲结婚后出生的兄弟姐妹,年龄分别为23岁、17岁、12岁及3岁。临床表现类似,运动发育落后及肌张力低下,均诊断为先天性肌营养不良。患者3、4及5病情缓慢加重,目前只能坐轮椅。检查发现轻度学习障碍,全身肌肉无力,近端肌无力更为明显,腱反射消失,双足有扁平足,腭弓高尖。血常规、电解质、肾功能及肝功能化验指标均正常。患者3及患者4脑核磁共振结果正常,呼吸机无力出现轻度呼吸困难,但没有严重到需要呼吸机治疗的地步,肌电图检查发现肌肉动作电位下降,肌纤维震颤增加。患者4在6岁时肌肉活检发现肌病样改变。患者6在15个月龄时表现为肌张力低下,腱反射消失,运动发育落后,不能站立,也不能爬行。所有患者ALG2基因检查均发现纯合突变(c.214_226delinsAGTCCCCGGC/p.72_75delinsSPR)。患者7为意大利人,父母为表兄妹近亲婚配,4岁起出现容易摔倒,摔倒后起身困难,爬楼梯吃力,被诊断为肌营养不良。10岁时症状加重,不能独立行走,吡啶斯的明治疗后症状好转,但仍然不能跑步,走路也需要帮助。目前60岁,只能独立行走15-20米,疲劳或感染时症状会加重。查体发现站立时摇晃,脊柱前凸明显,肩胛骨翼狀外翻,Gowers征明显(由仰卧位坐起时,有一个特征性的起立动作,即不能直接从仰卧位上坐起,需首先翻身成为俯卧位,然后再蹲起,再转换为四点支持位)。面部、眼睛及颈部肌肉力量正常,未见眼睑下垂。但四肢肌力降低,近端肌力降低尤为明显。肌电图检查发现肌肉复合动作电位下降到67%,单个肌纤维电位三分之二出现震颤或传导阻滞。肌肉活检发现非特异性肌损伤改变。基因检查发现ALG2基因纯合突变(c.203T4G,p.Val68Gly),功能学研究发现导致糖基化异常。2018年Asteggiano等报道2例ALG2-CDG患儿。患者8及患者9分别是8岁及10岁姐弟,父母为健康非近亲婚配家长。4个月龄起表现为难治性癫痫、运动发育落后及智力障碍等严重神经系统病变。特殊面容包括严重小头畸形、眼裂异常、鼻梁宽、双内眦赘皮及双眼内斜视。姐弟均有双眼虹膜缺损及单眼白内障。转铁蛋白等电聚焦电泳提示I型糖基化障碍,基因检查发现ALG2基因纯合突变(c.G752T/p.Arg251Leu)。患者8在8个月龄时检查头颅核磁共振发现基底节及脑室周围脑白质高信号,脑电图发现高度节律失常(提示婴儿痉挛症)。患者9头颅核磁共振胼胝体偏薄,幕上脑室系统扩张及额颞部蛛网膜下间隙增宽。2岁时出现脑室周围脑白质轻度异常,小脑半球信号增高,颞叶前段脑组织相对萎缩中度,侧脑室中度增大,脑电图提示高度节律失常。诊断及治疗:可通过基因检查、糖基化检测、神经肌肉系统检查及其他相关器官系统检查协助症状。治疗主要是对症支持治疗为主,乙酰胆碱酯酶抑制剂可能会减轻肌肉方面的异常。参考资料:1. https://www.cdghub.com/cdg/alg2/2. ThielC,SchwarzM,PengJ,GrzmilM,HasilikM,BraulkeT,KohlschütterA,vonFiguraK,LehleL,KörnerC.Anewtypeofcongenitaldisordersofglycosylation(CDG-Ii)providesnewinsightsintotheearlystepsofdolichol-linkedoligosaccharidebiosynthesis.JBiolChem.2003Jun20;278(25):22498-505.3. MoniesDM,Al-HindiHN,Al-MuhaizeaMA,JaroudiDJ,Al-YounesB,NaimEA,WakilSM,MeyerBF,BohlegaS.Clinicalandpathologicalheterogeneityofacongenitaldisorderofglycosylationmanifestingasamyasthenic/myopathicsyndrome.NeuromusculDisord.2014Apr;24(4):353-9.doi:10.1016/j.nmd.2013.12.010.Epub2014Jan4.PMID:24461433.4. CossinsJ,BelayaK,HicksD,SalihMA,FinlaysonS,CarboniN,LiuWW,MaxwellS,ZoltowskaK,FarsaniGT,LavalS,SeidhamedMZ;WGS500Consortium,DonnellyP,BentleyD,McGowanSJ,MüllerJ,PalaceJ,LochmüllerH,BeesonD.CongenitalmyasthenicsyndromesduetomutationsinALG2andALG14.Brain.2013Mar;136(Pt3):944-56.5. AsteggianoCG,PapazogluM,BistuéMillónMB,PeraltaMF,AzarNB,SpécolaNS,GuelbertN,SuldrupNS,PereyraM,DodelsondeKremerR.TenyearsofscreeningforcongenitaldisordersofglycosylationinArgentina:casestudiesandpitfalls.PediatrRes.2018Dec;84(6):837-841.
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科26人已读
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科26人已读 - 医学科普 FCSK(FUK)先天性糖基化障碍伴岩藻糖糖基化缺陷2型(FCSK-CDG或FUK-CDG)
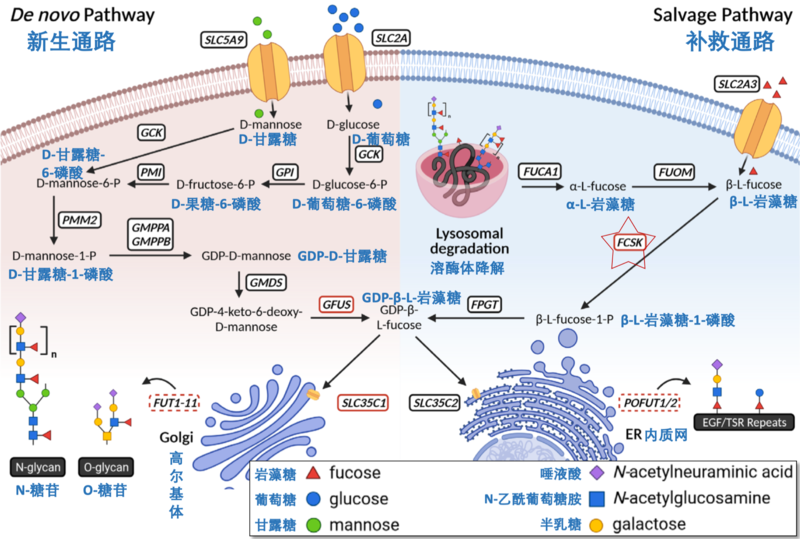
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生岩藻糖(Fucose)在决定ABO血型及路易斯血型等细胞内和细胞间识别当中至关重要。细胞膜上带有α-1,3-连接岩藻糖的多聚糖在白细胞和血管内皮细胞识别及粘附过程中可以启动白细胞驱动到炎症或感染组织。岩藻糖化的糖蛋白在哺乳动物受孕、胎儿发育及神经组织形成过程中必不可少。哺乳动物中,N糖基化及O糖基化过程均依赖于活化的岩藻糖(GDP岩藻糖)参与。GDP岩藻糖的合成依赖于新生通路(Denovopathway)及补救通路(salvagepathway)等两个途径。通常约90%的GDP岩藻糖合成来自新生通路的合成,以GDP甘露糖作为原料。约10%的GDP岩藻糖来自补救通路合成,以溶酶体中降解的L-岩藻糖作为原料,FCSK基因负责补救通路(salvagepathway)将L-岩藻糖转换为岩藻糖-1-磷酸,为最终形成的GDP岩藻糖提供原料。导致岩藻糖糖基化缺陷的疾病包括FCSK-CDG(FCSK先天性糖基化障碍)、GFUS-CDG(GFUS先天性糖基化障碍)、SLC35C1-CDG(SLC35C1先天性糖基化障碍)及FUT8-CDG(FUT8先天性糖基化障碍)【图1】。FCSK基因(又称FUK基因)突变导致的先天性糖基化障碍伴岩藻糖糖基化缺陷2型(FCSK-CDG)非常罕见,全世界仅报道数例。FCSK基因直接影响GDP岩藻糖合成的补救通路,引起岩藻糖糖基化缺陷,导致先天性糖基化障碍。FCSK-CDG可表现为发育落后、智力障碍、脑病、婴儿痉挛症、难治性癫痫及肌张力低下。也有喂养困难、胃食管返流、肠蠕动障碍,营养不良、坏死性小肠炎、肝功能异常、智力异常、脑核磁共振异常、反复呼吸道感染、眼底检查异常、视力异常、关节挛缩、轻度同型半胱氨酸及甲基丙二酸升高、早产、慢性腹泻、营养不良等表现。2018年报道的第一例患者为男性、因为肌张力低下、吞咽困难、胃食管返流及肠胃蠕动功能不好出现喂养困难,最终需要鼻饲喂养。就诊时存在严重的发育落后、智力障碍、肌张力低下、难治性癫痫及癫痫性脑病。脑电图检查提示癫痫波及背景活动异常提示癫痫及脑功能不好。脑核磁共振检查发现胼胝体形态异常、大脑天幕上脑室系统形态异常、脑白质髓鞘化延迟。患者出现反复呼吸道感染、呼吸暂停、有时候需要抢救治疗。眼科检查发现双眼对称性视网膜黄斑病变及严重的视力障碍。血串联质谱检查可见持续同型半胱氨酸升高(18.2–22.3,正常范围4–14mmol/L)及甲基丙二酸升高(0.94–1.2,正常范围0–0.5nmol/L),补充维生素B12后同型半胱氨酸及甲基丙二酸水平稍有下降。维生素B12治疗之前血维生素B12水平偏高,而且维生素B12治疗之后同型半胱氨酸及甲基丙二酸水平均未降到正常水平,说明维生素B12缺乏不是其原因。2018年报道的第二例患者为25周早产的女性患儿,有喂养困难、呼吸困难、发育落后、严重智力障碍、难治性癫痫及肌张力低下。喂养困难表现为经口喂养时呛奶,需要胃管喂养。曾经因为坏死性小肠炎进行肠道切除治疗,之后出现营养吸收不良及腹泻。但早期治疗后营养状况改善。脑核磁共振检查提示小脑萎缩、胼胝体发育不良、脑室旁脑白质软化、囊性脑软化及脑桥小脑发育不良。癫痫刚开始表现为婴儿痉挛症及高度节律失常的脑电图改变,后来演变为眨眼动作及行为停止。眼部表现包括斜视、视神经萎缩及皮质性失明。血串联质谱表现类似于第一例患者。2022年报道的索马里女孩为11月龄,表现为肌张力低下、腱反射亢进、不能独坐、发育落后及失明。早在三个月龄时父母发现孩子不能追视。左侧肩胛骨外侧可见1.5x1.0cm大小的牛奶咖啡斑。脑核磁共振检查提示双侧额叶、顶叶及枕叶脑室周围脑白质T2高信号、侧脑室扩张、顶叶和颞叶脑组织软化灶。2022年报道的4.5岁伊朗男孩,婴儿期有过呼吸困难、喂养困难、胃管喂养、营养不良、发育落后及肌张力低下。之前有追视,但视力逐渐变差,到了两岁时失明。眼科检查发现对称性视网膜黄斑病变及视力障碍。3岁时出现强直痉挛性抽搐及癫痫,但丙戊酸钠治疗后得以控制。脑核磁共振检查提示小脑及小脑蚓部发育不良及胼胝体发育不良。4.5岁时语言发育落后,无法说出有意义的词语,还有智力障碍(IQ<50)。此外患者出现四肢关节挛缩,无法完成站立、行走或独坐等任务。2023年报道的3岁沙特男孩,6个月龄时被母亲发现眼神异常及抽动,检查发现高度节律失常的脑电图表现及婴儿痉挛症、抗癫痫治疗3个月后得以控制。3岁时患儿可以跑步、爬楼梯、骑三轮车,也能单腿站立片刻,语言功能及神经系统检查未见异常。治疗:FCSK-CDG是一类合并有岩藻糖化缺陷的先天性糖基化障碍,岩藻糖化缺陷的先天性糖基化障碍还包括GFUS-CDG、SLC35C1-CDG及FUT8-CDG。有报道此类疾病长期补充L-岩藻糖治疗后可以改善患者白细胞升高、反复感染、发育落后、矮小、营养不良等异常表现。理论上来说补充甘露糖可能会促进GDP岩藻糖合成的新生通路,改善岩藻糖化过程,从而改善患者临床症状。参考资料:1. NgBG,RosenfeldJA,EmrickL,JainM,BurrageLC,LeeB;UndiagnosedDiseasesNetwork;CraigenWJ,BeardenDR,GrahamBH,FreezeHH.PathogenicVariantsinFucokinaseCauseaCongenitalDisorderofGlycosylation.AmJHumGenet.2018Dec6;103(6):1030-1037. 2. HüllenA,FalkensteinK,WeigelC,HuidekoperH,Naumann-BartschN,SpengerJ,FeichtingerRG,SchaefersJ,FrenzS,KotlarzD,MomenT,KhoshnevisanR,RiedhammerKM,SanterR,HergetT,RenningsA,LefeberDJ,MayrJA,ThielC,WortmannSB.Congenitaldisordersofglycosylationwithdefectivefucosylation.JInheritMetabDis.2021Nov;44(6):1441-14523. ÖzgünN,ŞahinY.Acasewithcongenitaldisorderofglycosylationwithdefectivefucosylation2andnewmutationinFUKgene.BrainDev.2022Mar;44(3):239-243. 4. ManoochehriJ,KamalN,KhamiraniHJ,ZoghiS,HaghighiMF,GoodarziHR,BagherTabeiSM.AcombinationoftwonovelshomozygousFCSKvariantscausedisorderofglycosylationwithdefectivefucosylation:Newpatientandliteraturereview.EurJMedGenet.2022Aug;65(8):104535. 5. AlTuwaijriA,AlyafeeY,UmairM,AlsubaitA,AlharbiM,AlEidiH,BallowM,AldreesM,AlamQ,AlAbdulrahmanA,AlrifaiMT,AlfadhelM.Congenitaldisorderofglycosylationwithdefectivefucosylation2(FCSKgenedefect):Thethirdreportintheliteraturewithamildphenotype.MolGenetGenomicMed.2023Apr;11(4):e2117.
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科39人已读
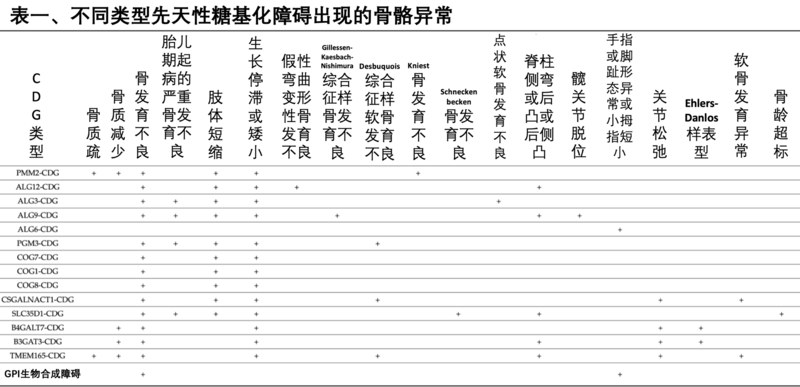
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科39人已读 - 医学科普 先天性糖基化障碍患者骨骼发育异常、骨密度降低及相关基因
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生先天性糖基化障碍(CDG)是累及多器官系统的遗传病。糖基化过程在骨发育、软骨发育及骨代谢当中至关重要,先天性糖基化障碍可以导致骨骼发育异常及骨密度降低。目前已知 ALG12先天性糖基化障碍(ALG12-CDG)、ALG3先天性糖基化障碍(ALG3-CDG)、ALG9先天性糖基化障碍(ALG9-CDG)、ALG6先天性糖基化障碍(ALG6-CDG)、PGM3先天性糖基化障碍(PGM3-CDG)、CSGALNACT1先天性糖基化障碍(CSGALNACT1-CDG)、SLC35D1先天性糖基化障碍(SLC35D1-CDG)以及TMEM165先天性糖基化障碍(TMEM165-CDG)等先天性糖基化障碍类型均明显导致骨骼发育不良。部分患者胎儿期即出现严重的骨骼发育不良,也可能会导致早期夭折。几乎所有CDG类型均可发现不同程度的骨质疏松或骨密度降低,而且到了成人期更为明显。骨密度降低的常见原因可能是激素紊乱、骨骼发育异常、骨代谢紊乱、运动障碍或减少以及营养物质缺乏。因为患者、家长和医护人员认识不足,先天性糖基化障碍患者骨骼异常容易被低估或被忽略。为了避免骨折发生或慢性疼痛,不仅需要对骨骼病变有高度警惕性,而且需要详细评估治疗CDG患者骨骼异常。随着CDG治疗手段不断增多,有望减少骨骼病变或降低骨骼异常导致的并发症。此文详细介绍CDG患者骨骼异常的表现、可能的原因、骨质疏松高危因素和需要监测的骨骼或骨代谢指标。PMM2先天性糖基化障碍患者出现的骨骼异常PMM2先天性糖基化障碍是最常见的CDG,可通过内分泌异常、IGFBP3功能障碍以及IGF通路异常导致矮小症,故生长障碍在PMM2-CDG患者中常见。虽然血钙、血磷和血镁水平正常,儿童期即经常出现骨质疏松或骨密度降低。虽然经常被忽视,其他骨骼异常也常见,包括脊柱侧弯及脊柱后凸,可导致脊髓受压、脊柱压缩或骨折。如果脊柱侧弯或后凸明显,需要定期骨科评估。骨折比较常见,而且看似可以正常愈合。骨骼发育不良虽然不常见,但也被报道过。关节挛缩比较常见,而且可以影响患者生活质量。其他糖基化障碍出现的骨骼发育不良跟PMM2先天性糖基化障碍(PMM2-CDG)相比,其他CDG类型的患者骨骼发育不良相对常见,包括ALG12-CDG,、ALG3-CDG、ALG9-CDG、ALG6-CDG、PGM3-CDG、CSGALNACT1-CDG、SLC35D1-CDG 和TMEM165-CDG。ALG12先天性糖基化障碍(ALG12-CDG)患者发现的骨骼异常包括指间关节脱位、马蹄内翻足、近侧肢体缩短、中脸发育不良、掌骨缩短、水平位的髋臼脊及脊柱侧弯。大部分PGM3-CDG患者存在骨骼异常,其中脊柱侧弯是轻度的骨骼异常。而2例患者存在严重的Desbuquois 骨发育不良,表现为严重的胎儿期及出生后发育落后、矮小症、四肢短小、长骨缩短、干骺端杯口状改变、股骨小粗隆肥大、骨龄超标、关节松弛及进行性加重的脊柱后侧凸。COG7-CDG、COG1-CDG及COG8-CDG 均已报道存在骨骼发育不良作为其主要临床表现。COG8-CDG的骨发育不良严重,而COG1-CDG、COG7-CDG患者骨发育不良较轻微。先天性糖基化障碍患者关节及软骨异常2例SLC35D1-CDG患者发现典型的 Schneckenbecken骨发育不良(软骨发育不良合并有蜗牛样盆骨),是胎儿期出现的致命性骨发育不良,髂骨看似像蜗牛、胸廓发育不良、长骨变短变粗,也会有椎体扁平。B4GALT7-CDG患者发现Ehlers-Danlos 综合征(又称埃勒斯-当洛综合征或过度弹性皮肤综合征)样骨发育不良,包括桡尺骨融合、全身性骨密度降低、肋骨外翻、拇指偏宽、手指偏长、脚趾偏长而且重叠、马蹄内翻足、横断掌及关节松弛。此外,TMEM165-CDG报道矮小症、全身性骨密度降低或骨质疏松,骨骺和干骺端发育不良。GPI生物合成障碍导致的先天性糖基化障碍患者出现的骨骼异常GPI生物合成障碍导致的先天性糖基化障碍包括DPM1-CDG、DPM2-CDG、DPM3-CDG、MPDU1-CDG、PIGA-CDG、PIGB-CDG、PIGC-CDG、PIGF-CDG、PIGG-CDG、PIGH-CDG、PIGL-CDG、PIGM-CDG、PIGN-CDG、PIGO-CDG、PIGP-CDG、PIGQ-CDG、PIGV-CDG、PIGW-CDG、PIGX-CDG、PIGY-CDG、GPA1-CDG、MPPE1-CDG、PGAP1-CDG、PGAP2-CDG、PGAP3-CDG、PIGK-CDG、PIGS-CDG、PIGT-CDG及PIGU-CDG。上述CDG类型具有独特的骨骼系统异常表现,包括手指或脚趾形态异常、远端指骨或趾骨缺失、手指或脚趾发育不良、手指或脚趾短小、手指或脚趾偏宽、杵状指、指弯曲及骨密度降低等。所有PIGV-CDG患者及大部分PIGO-CDG患者均报道存在远端指骨短小。表一总结了不同类型先天性糖基化障碍出现的骨骼异常。CDG患者出现骨质疏松的高危因素促性腺激素不足性腺功能低下(hypogonadotrophichypogonadism)导致的激素紊乱是女性CDG患者骨密度降低的危险因素,可能需要雌激素替代治疗来预防骨质疏松。但雌激素治疗可能会增加血栓形成的风险,需要谨慎调整激素用量。严重突变或严重影响骨代谢及骨发育的先天性糖基化障碍类型可以出现严重或致命性的骨骼异常。胃肠道的问题及营养吸收障碍可能会引起维生素D缺乏,值得监测25羟维生素D3水平,必要时加强补充维生素D3预防佝偻病。CDG患者中常见的骨骼畸形、慢性疼痛、神经系统异常引起的运动障碍或运动发育落后、坐轮椅、消瘦、肌萎缩等因素均可导致限制运动,引起骨密度降低或反复骨折。骨健康标记物骨形成的标记物:血清总碱性磷酸酶骨碱性磷酸酶血清骨钙素血清C1NP(1型前胶原C端)血清P1NP(1型前胶原N端)骨吸收或溶解的标记物:尿羟脯氨酸尿总的吡啶啉(PYD)尿游离脱氧吡啶啉(DPD)尿胶原1型交联N端肽(NTX)尿或血清1型交联C端肽(CTX)骨唾液酸蛋白(BSP)抗酒石酸酸性磷酸酶5b血钙、血磷、甲状腺功能、甲状旁腺功能、25羟维生素D3水平。碱性磷酸酶升高,尤其是骨碱性磷酸酶反映成骨细胞的活动。PMM2先天性糖基化障碍患者可以在有条件的机构检测血清及尿液脱氧吡啶啉(deoxypyridinoline)、吡啶啉(pyridinoline)、稳定细胞外基质的交联氨基酸等标记物来筛查骨密度减少症。CDG患者中需要进一步研究确定骨骼形成的标记物(P1NP)及骨骼吸收的标记物(CTX)应用价值。所有的骨骼标记物均应该考虑症状、年龄、性别、运动能力及饮食等因素后综合判断期意义。转铁蛋白糖基化检测及N糖苷检测可以反映糖基化异常是否改善,用于监测疗效。但能否用于监测骨骼异常改善程度有待进一步研究。骨密度检测可用于评估骨质疏松是否存在及严重程度,也可以用于监测治疗的效果。全脊柱X光平片可以用于发现骨骼畸形、压缩或骨折。骨骼的定量CT扫描(QCT)是定量评估骨密度的敏感措施。体质成份分析越来越被用于评估遗传代谢病患者当中,能够提供骨骼、脂肪及肌肉等组织方面的参考信息。针对骨骼健康的治疗措施:PGM1先天性糖基化障碍(PGM1-CDG)及TMEM165先天性糖基化障碍(TMEM165-CDG)等CDG存在N糖苷半乳糖化不足,补充半乳糖后治疗效果良好。UDP半乳糖转运酶异常导致的SLC35A2先天性糖基化障碍(SLC35A2-CDG)虽然补充半乳糖后糖基化异常得以改善,能否改善骨骼病变仍需要进一步被证实。甘露糖治疗可以改善MPI先天性糖基化障碍(MPI-CDG)患者内分泌紊乱、凝血异常和肠病等表现,仍需进一步研究对长期治疗对骨骼系统异常的疗效。 UDP岩藻糖转运酶异常导致的SLC35C1先天性糖基化障碍(SLC35C1-CDG)出现高尔基体的岩藻糖化异常,补充岩藻糖可以改善病情。 虽然以上治疗措施可以改善患者血液和化验指标,对骨骼系统的治疗作用病不明确,需要长期观察研究食物补充剂治疗及其他新的治疗措施对CDG患者骨骼健康的疗效。总结骨发育不良的患者应该筛查有没有先天性糖基化障碍,尤其是合并有其他器官系统异常的时候更应该重点筛查有没有CDG。胎儿期出现骨发育不良的患者预后可能不好,故需要早期基因诊断以便进行遗传咨询或遗传筛查。因为CDG患者骨骼病变常被忽略或低估,确诊或怀疑CDG时需要有高度的警惕性并且仔细评估骨骼病变。成人或青少年患者中应该筛查骨质疏松及其高危因素。骨代谢指标的监测以及对应的诊治措施有望改善患有骨骼异常的CDG患者病情或预防严重骨骼并发症的出现。参考资料:1. LipińskiP,StępieńKM,CiaraE,Tylki-SzymańskaA,Jezela-StanekA.SkeletalandBoneMineralDensityFeatures,GeneticProfileinCongenitalDisordersofGlycosylation:Review.Diagnostics(Basel).2021Aug9;11(8):1438. 2. https://emedicine.medscape.com/article/128567
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科30人已读
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科30人已读 - 医学科普 STT3A先天性糖基化障碍(STT3A-CDG)及STT3B先天性糖基化障碍(STT3B-CDG)
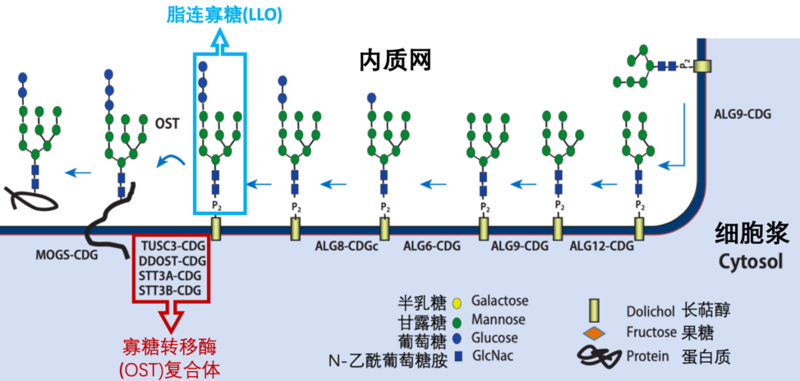
复旦大学附属儿科医院肝病科,库尔班江·阿布都西库尔医生蛋白糖基化的过程中,内质网里连接在长萜醇的脂连寡糖(LLO)需要被转移到蛋白质的,这一过程是通过寡糖转移酶(OST)复合体来完成的。人类当中OST复合体包括含有STT3A的OST-A和含有STT3B的OST-B两种多聚(multimeric) 蛋白。人类OST-A和OST-B复合体均还包括六个蛋白:核糖体亲和蛋白I(ribophorin-I)、核糖体亲和蛋白II、DDOST(又称OST48)、TMEM258、DAD1及OST4。OST-A还包括KRTCAP2及OSTC,而OST-B还包括MAGT1或TUSC3。近年来相继发现OST复合体相关基因突变导致的先天性糖基化障碍,包括DDOST-CDG(又称OST48-CDG)、TUSC3-CDG、MAGT1-CDG 、STT3A-CDG及STT3B-CDG【图1】。此文详细描述STT3A先天性糖基化障碍(STT3A-CDG)及STT3B先天性糖基化障碍(STT3B-CDG)的特点。 STT3A先天性糖基化障碍(STT3A-CDG)STT3A先天性糖基化障碍(STT3A-CDG)分为常染色体显性遗传的STT3A先天性糖基化障碍(STT3A-CDG)(STT3A基因单个杂合新发突变或来自父母一方的突变)及常染色体隐性遗传的STT3A先天性糖基化障碍(STT3A-CDG)(STT3A基因两个突变,分别来自父母)。1、常染色体显性遗传的STT3A先天性糖基化障碍(STT3A-CDG)2021年欧美多国科学家报道16例常染色体显性遗传的STT3A先天性糖基化障碍(STT3A-CDG)患者。16例患者疾病表现多样:10例患者发现骨骼异常或骨骼发育不良,9例患者有智力障碍,8例患者存在矮小症、6例小头畸形,5例存在肌张力升高、4例患者存在早发型关节炎及乳头内陷,3例患者脂肪异常分布,2患者伴有宫内发育不良、肌张力低下及球形红细胞增多症。也有患者发现腓肠肌肥大、肌肉痉挛或抽筋【图二】。报道的16例患者中男女各占一半,最小年龄3岁,最大年龄55岁,10例患者存在不同程度的发育落后或智力障碍。所有患者足月出生,2例患者有宫内发育不良(出生体重不达标),6例患者大头畸形但2例患者存在进行性加重的小头畸形。大部分患者特殊面容表现轻微,包括发际前缘偏低、眼裂小、上唇薄、宽鼻梁及下巴前凸。4例患者可见招风耳及乳头内陷,3例儿童脂肪异常分布(臀部周围脂肪垫),一例儿童毛发增多。儿童患者长到青年期之前通常是健康的,6例儿童2-3岁时才会走路,存在轻到中度的运动发育落后。1例儿童运动发育落后严重,存在肌阵挛及共济失调(平衡能力差),后来检查发现视神经萎缩、斜视、痉挛性双瘫、后颅窝增大及蛛网膜囊肿。5例患者肌张力升高,2例肌张力低下。7例儿童存在语言发育落后,其中2例没有出现语言的发育(始终不能说话)。有一例患者10岁时出现肥胖症,体重指数29.1(第99个百分位点)。2例患者存在球形红细胞增多症。10例患者存在骨骼异常,包括3例长骨干骺端的轻度杯口状改变(metaphysealflaring)及骨骺端异常,2例脊柱异常,一例胫骨及尺骨骨骺端畸形及脊柱侧弯。3例患者囟门闭合延迟,4例成人30岁开始既有关节炎的早期表现直到40岁需要双侧髋关节置换手术。7例患者出现肌肉痉挛或疼痛。8例患者存在矮小症。9例患者发现智力障碍,一例成人患者存在轻微学习障碍但没有智力低下。所有患者看到的胃肠道表现轻微,但没有看到心脏、肾脏或肺部等部位的异常。常规化验很少发现异常,转氨酶水平正常,2例患者发现凝血因子活性降低,1例患者发现雄激素水平下降,另外1例发现促甲状腺素水平升高但没有甲状腺功能减低的临床症状。2例出现青春发育期延后,另一例接受生长激素治疗。图三、常染色体显性遗传的STT3A先天性糖基化障碍(STT3A-CDG)患者发现的异常:(A)乳头内陷、腿短、脊柱前凸、脊柱侧弯、翼状肩胛骨及腓肠肌肥大。(B)鼻梁宽、发际低、鼻尖窄及弓状眉毛。(C)鼻梁宽、颧骨狭窄、发际低、鼻梁凸出、鼻尖窄、耳廓偏低、眼窝凹陷及弓状眉毛。(D和E)大头畸形、发际前缘低、耳廓偏低及中脸后缩;弟弟眼裂下斜、短鼻及帐篷状上唇。(F)大头畸形、前发际偏高、额头窄、中脸后缩、短鼻、小下颌、耳廓偏低且后转、口角下垂。(G)16岁时骨骼片提示骶髂部昆虫骨片形成或硬化、脊柱侧弯、胫骨弯曲、尺骨形态不规则及2-3掌骨偏大。(H)2岁时T1脑核磁共振信号和12岁时T2脑核磁共振检查:幕上及大脑半页间非进行性肿大的蛛网膜囊肿及后窝稍大。2、常染色体隐性遗传的STT3A先天性糖基化障碍(STT3A-CDG)常染色体隐性遗传的STT3A先天性糖基化障碍(STT3A-CDG)可以出现发育落后、智力障碍、营养不良、小头畸形、抽搐、发育落后、视神经萎缩及抽搐。一例患者发现凝血因子VIII和血管性血友病因子(vonWillebrandFactor)活性降低2013年Shrimal等报道一例巴基斯坦裔近亲婚配家庭常染色体隐性遗传的STT3A先天性糖基化障碍(STT3A-CDG)患者,表现为发育落后、智力障碍、小脑萎缩、小头畸形、喂养困难(需要鼻饲喂养)、营养不良、肌张力低下及抽搐。13岁时不能独坐,追视差,而且有难治性癫痫。糖基化筛查提示I型糖基化障碍,基因检测发现STT3A基因c.1877T>C/p.Val626Ala纯合突变,分别来自父母。功能学研究发现此突变导致蛋白表达及糖基化异常。STT3B先天性糖基化障碍(STT3B-CDG)目前为止发现的病例均为常染色体隐性遗传模式,患者1个纯合突变,父母均为基因突变的杂合携带者。常染色体隐性遗传模式也可以因为患者携带两个分别来自父母的杂合突变,父母为健康杂合携带者,父母可以不是近亲婚配。2020年Kılıç等报道土耳其裔近亲婚配家庭出生的STT3B先天性糖基化障碍(STT3B-CDG)孩子。胎儿期发现胎动减少及宫内发育不良,怀孕38周,出生体重仅2660g(不达标) ,身高47cm(不达标) ,头围34cm(不达标) 。3个月龄时发现运动发育落后、特殊面容、肌张力低下及腱反射减弱。眼底检查发现视乳头轻度苍白(minorpalepapillae),视觉诱发电位异常,但听觉诱发电位正常。1岁时出现抽搐并接受苯二氮卓类药物治疗。5岁时因呼吸道感染就诊时发现有特殊面容(毛发增多、眼裂小、额头窄、眼距增宽、长睫毛、外眼角上翘)、躯干部肌张力低下、脂肪分布异常(脂肪垫)、肌力减弱及进行性加重的发育落后。因为呼吸暂停反复发作,需要长期吸氧治疗,后来需要气管切开进行人工呼吸。化验检查仅发现肾功能指标及转氨酶短暂异常,B超发现双肾肾盂轻度扩张。脑电图检查可见爆发-抑制脑电波,脑核磁共振可见弥漫性髓鞘化延迟、第三脑室和第四脑室扩张、侧脑室周围胶质增生改变。转铁蛋白糖基化筛查结果正常,但基因检查发现STT3B基因c.38C>G/p.Ser13Trp纯合突变,父母均为杂合携带者。2013年Shrimal等报道一例伊拉克裔近亲婚配家庭出生的STT3B先天性糖基化障碍(STT3B-CDG)孩子。表现为宫内发育不良、小头畸形、营养不良、发育落后、喂养困难(需要鼻饲喂养)、呼吸困难、智力障碍、小脑萎缩、癫痫、不能追视、视神经萎缩、肌张力低下、肝功能异常、血小板减少、生殖器异常(小阴茎、阴囊发育不良及隐睾)。患者4岁时过世。过世前糖基化筛查发现轻微糖基化异常,基因检测发现STT3B基因c.1539+20G>T纯合突变,功能分析发现蛋白表达严重减少。参考资料:1. WilsonMP,GarantoA,PintoEVairoF,NgBG,RanatungaWK,VentouratouM,BaerenfaengerM,HuijbenK,ThielC,AshikovA,KeldermansL,SoucheE,Vuillaumier-BarrotS,DupréT,MichelakakisH,FiumaraA,PittJ,WhiteSM,LimSC,GallacherL,PetersH,RymenD,WittersP,RibesA,Morales-RomeroB,Rodríguez-PalmeroA,BallhausenD,deLonlayP,BaroneR,JanssenMCH,JaekenJ,FreezeHH,MatthijsG,MoravaE,LefeberDJ.ActivesitevariantsinSTT3AcauseadominanttypeIcongenitaldisorderofglycosylationwithneuromusculoskeletalfindings.AmJHumGenet.2021Nov4;108(11):2130-2144.doi:10.1016/j.ajhg.2021.09.012.Epub2021Oct14.PMID:34653363;PMCID:PMC8595932.2. ChangIJ,ByersHM,NgBG,MerrittJL2nd,GilmoreR,ShrimalS,WeiW,ZhangY,BlairAB,FreezeHH,ZhangB,LamC.FactorVIIIandvWFdeficiencyinSTT3A-CDG.JInheritMetabDis.2019Mar;42(2):325-332.doi:10.1002/jimd.12021.Epub2019Jan30.Erratumin:JInheritMetabDis.2019May;42(3):578.PMID:30701557;PMCID:PMC6658093.3. ShrimalS,NgBG,LosfeldME,GilmoreR,FreezeHH.MutationsinSTT3AandSTT3Bcausetwocongenitaldisordersofglycosylation.HumMolGenet.2013Nov15;22(22):4638-45.doi:10.1093/hmg/ddt312.Epub2013Jul10.PMID:23842455;PMCID:PMC3888133.4. GhoshA,UrquhartJ,DalyS,FergusonA,ScotcherD,MorrisAAM,Clayton-SmithJ.PhenotypicHeterogeneityinaCongenitalDisorderofGlycosylationCausedbyMutationsinSTT3A.JChildNeurol.2017May;32(6):560-565.doi:10.1177/0883073817696816.Epub2017Mar16.PMID:28424003.5. KılıçB,AkkuşN.NovelmutationandsevererespiratoryfailureincongenitaldisordersofglycosylationTypeIx.TurkJPediatr.2020;62(1):114-118.doi:10.24953/turkjped.2020.01.016.PMID:32253875.
 库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科35人已读
库尔班江·阿布都西库尔 副主任医师 复旦大学附属儿科医院 肝病科35人已读

库尔班江·阿布都西库尔副主任医师
复旦大学附属儿科医院肝病科